Decoding Variant Classification: A Comprehensive Guide to ACMG AMP Criteria for Researchers & Drug Developers
This article provides researchers, scientists, and drug development professionals with an in-depth analysis of the ACMG (American College of Medical Genetics and Genomics) and AMP (Association for Molecular Pathology) joint...
Decoding Variant Classification: A Comprehensive Guide to ACMG AMP Criteria for Researchers & Drug Developers
Abstract
This article provides researchers, scientists, and drug development professionals with an in-depth analysis of the ACMG (American College of Medical Genetics and Genomics) and AMP (Association for Molecular Pathology) joint consensus guidelines for variant interpretation. It explores the foundational framework, methodological application in genomic analysis, common challenges and optimization strategies, and comparative validation against other systems. The content serves as a critical resource for ensuring standardized, evidence-based variant classification in research pipelines and therapeutic development.
The ACMG-AMP Blueprint: Understanding the Foundational Framework for Variant Interpretation
This document provides a detailed historical analysis and technical protocol guide for the evolution of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) variant interpretation guidelines from their 2015 inception to present-day refinements. Framed within a broader thesis on the standardization of genomic medicine, these notes are designed to equip researchers and drug development professionals with the contextual understanding and practical methodologies necessary for robust variant classification in both clinical diagnostics and therapeutic target validation.
Historical Context and Quantitative Evolution (2015-Present)
The 2015 ACMG/AMP publication established a seminal, semi-quantitative framework for classifying sequence variants into five categories: Pathogenic (P), Likely Pathogenic (LP), Variant of Uncertain Significance (VUS), Likely Benign (LB), and Benign (B). This framework leveraged 28 criteria weighted by evidence type (Population, Computational, Functional, Segregation, De novo, Allelic, Other Database, Other). Subsequent updates have addressed its limitations, enhancing reproducibility and specificity for various gene contexts.
Table 1: Evolution of ACMG/AMP Guidelines: Key Publications and Impact
| Year | Key Publication/Update | Primary Focus & Evolution | Impact on Classification Consistency |
|---|---|---|---|
| 2015 | Richards et al. (Genet Med) | Original framework with 28 criteria (16 for pathogenicity, 12 for benignity). | Established baseline standard; inter-laboratory variability remained high. |
| 2018 | ClinGen SVI Recommendations | Standardized application of PS1/PM5 (same amino acid change) and PS3/BS3 (functional assays). | Reduced subjective weighting; introduced calibrated approaches for functional data. |
| 2019-2020 | ClinGen Gene-Disease Curation | Introduced Clinical Validity Curation (Definitive, Strong, Moderate, Limited) to inform PVS1 strength. | Enabled gene-specific modification of criteria strength (e.g., PVS1 attenuation for non-loss-of-function mechanisms). |
| 2020 | ClinGen/CAP Variant Interpretation Guidelines | Focused on copy number variants (CNVs) and secondary findings. | Extended framework beyond single nucleotide variants/small indels. |
| 2021-2023 | ACMG/AMP ClinGen Revision (v3.0) | Refined, redefined, and added new criteria. Major changes: Re-evaluation of PP2/BP1; new PP5/BP6; introduction of Re codes for curated assertions. | Addressed circular logic, improved transparency, and formally integrated public data sharing. |
| 2024-Present | Ongoing ClinGen Expert Panels | Development of gene- and disease-specific specifications (e.g., for TP53, PTEN, CDH1, MYH7). | Significantly reduces VUS rates and improves clinical actionability for specific conditions. |
Table 2: Summary of Key Criterion Modifications (2015 vs. Post-2021)
| Criterion | 2015 Original Description | Post-2021 Key Refinements |
|---|---|---|
| PVS1 | Null variant in a gene where LOF is a known mechanism. | Stratified strength (PVS1VeryStrong to PVS1_Moderate) based on location (e.g., initiating Met, nonsense-mediated decay status) and gene-disease mechanism. |
| PS3/BS3 | Well-established functional studies. | Requires use of ClinGen-approved clinical domain-specific functional frameworks for calibration (e.g., for missense variants). |
| PM2 | Absent from population databases. | Thresholds and population stratification explicitly defined using gnomAD v3.0+ allele frequency data. |
| PP2/BP1 | Missense variant in a gene with low rate of benign missense. | More stringent application; requires statistical support from missense constraint metrics (e.g., missense Z-score >3.09). |
| PP5/BP6 | Reputable source without data. | Deprecated in 2015 form. New PP5 is for computational evidence with high prediction scores; BP6 is for population frequency above disease prevalence. |
Experimental Protocols for Key Evidence Types
Protocol 3.1: Application of PM2 (Population Data Curation)
- Objective: To quantitatively apply the PM2 (Absent/Extremely Low Frequency) criterion using current population genomic databases.
- Materials: High-performance computing terminal, access to gnomAD (v4.0+), and disease-specific prevalence data.
- Methodology:
- Extract the allele frequency (AF) for the variant from the latest gnomAD genome (v4.0) and exome datasets, noting population-specific frequencies.
- Calculate the maximum credible population allele frequency for the disease phenotype using the formula: Prevalence / (2 * Penetrance). Use the highest plausible population prevalence estimate.
- Apply PM2Supporting: If the total AF is < 0.0005 (0.05%) and the variant is absent in the subpopulation most relevant to the patient.
- Apply PM2Moderate: If the total AF is < 0.00002 (0.002%) and meets the above condition.
- Apply PM2: If the total AF is less than the calculated maximum credible allele frequency for a dominant disorder (or appropriate multiple for recessive).
- Note: For recessive disorders, apply PM2 in trans with a known pathogenic variant.
Protocol 3.2: Calibrated Functional Assay Application (PS3/BS3)
- Objective: To evaluate experimental data for classification using the ClinGen Sequence Variant Interpretation (SVI) Functional Assay Working Group framework.
- Materials: Published or internally generated functional study data, calibration thresholds from relevant ClinGen Expert Panel specifications.
- Methodology:
- Assay Classification: Determine if the assay is Definitive, Strong, Moderate, or Supporting based on its ability to recapitulate the biological pathway and its validated positive/negative controls.
- Data Extraction: Quantify the variant's functional impact (e.g., % residual activity, fold-change in protein expression, localization score) relative to wild-type and known pathogenic/benign controls.
- Calibration: Apply the pre-defined thresholds from the relevant gene/disease specification. Example for a Definitive assay: >80% wild-type activity → BS3Strong; <20% wild-type activity → PS3Strong.
- Integration: Combine the assay strength (Step 1) with the observed effect size (Step 3) to assign the final criterion strength (e.g., PS3Moderate, BS3Supporting).
Protocol 3.3: Gene-Specific PVS1 Application
- Objective: To stratify the strength of PVS1 based on variant type and gene-disease mechanism.
- Materials: Transcript annotation tools (e.g., VEP, SpliceAI), ClinGen Gene-Disease Clinical Validity and LOF Curation reports.
- Methodology:
- Establish the Gene-Disease Mechanism: Consult ClinGen curation to confirm LOF is an established disease mechanism (Definitive/Strong evidence).
- Variant Annotation:
- Identify canonical transcript and protein impact.
- For nonsense variants: Predict nonsense-mediated decay (NMD) likelihood. Variants in the last exon or >50-55 nucleotides upstream of the last exon-exon junction are often NMD-escape.
- Stratification (Per SVI Recommendations):
- PVS1: Null variant (nonsense, frameshift, canonical ±1/2 splice) in a gene where LOF is the sole known mechanism, and variant is predicted to undergo NMD.
- PVS1Strong: Same as above, but in a gene where LOF is not the sole known mechanism.
- PVS1Moderate: Variant is predicted to escape NMD (e.g., last exon stop-loss).
- PVS1_Supporting: Non-canonical splice site variant with high likelihood of LOF.
Visualizations
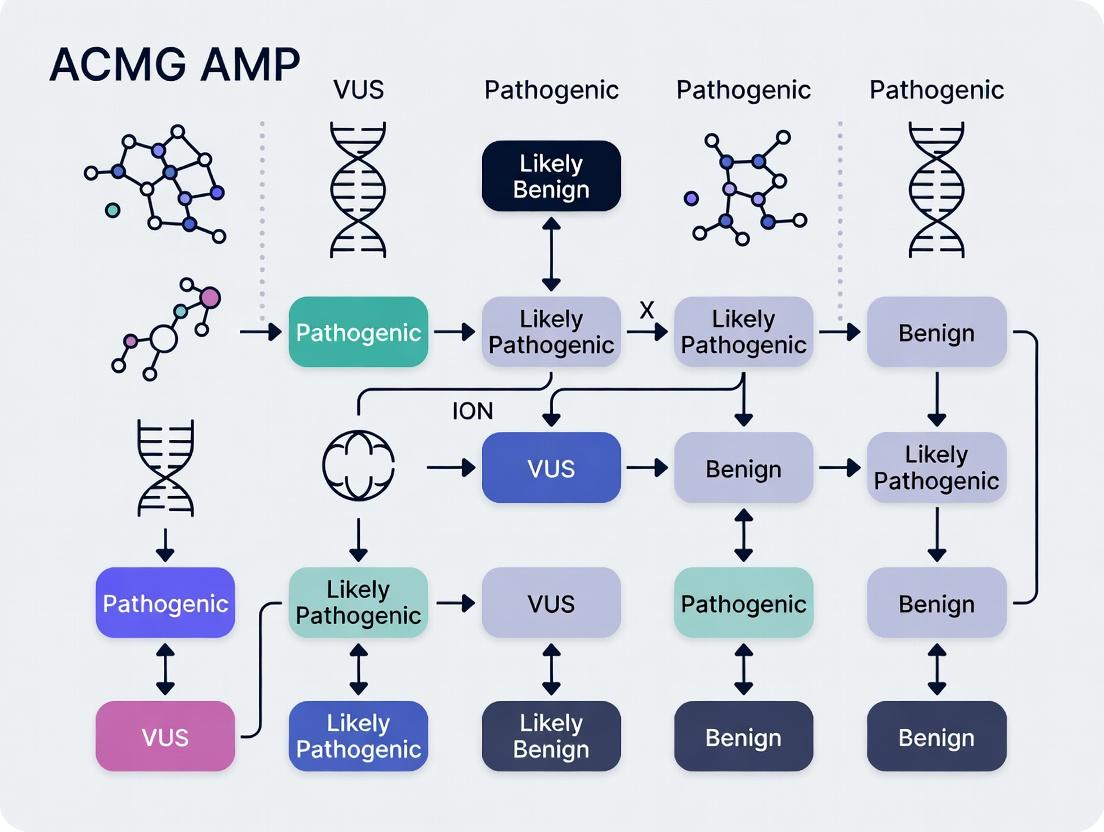
Diagram 1: ACMG/AMP Variant Classification Workflow (2024)
Diagram 2: Evolution of Key Criterion PS3/BS3 Application
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Variant Interpretation & Functional Validation
| Reagent/Tool | Provider/Example | Function in ACMG/AMP Research |
|---|---|---|
| Reference Genomes & Annotations | GRCh38/hg38, GENCODE, RefSeq | Standardized genomic coordinates and transcript definitions for consistent variant annotation (critical for PVS1, PM4). |
| Population Frequency Databases | gnomAD, TOPMed, UK Biobank | Provides allele frequency data for applying BA1, BS1, BS2, PM2, and PM3 criteria. |
| In silico Prediction Suites | REVEL, MetaLR, SpliceAI, AlphaMissense | Computational evidence for PP3 (supporting pathogenic) and BP4 (supporting benign) criteria. |
| Clinically Curated Variant Databases | ClinVar, LOVD, HGMD (subscription) | Source of other database evidence (PS4, PM5, PP5 legacy) and literature associations. |
| Gene Constraint Metrics | gnomAD pLI & missense Z-score | Informs application of PP2 and BP1; genes with high missense Z-score (>3.09) are more tolerant. |
| Functional Assay Kits (e.g., Splicing Reporters) | Minigene construction kits (pSPL3, pCAS2) | Experimental validation of splice-altering variants for PS3/BS3 evidence. |
| Plasmid Mutagenesis Kits | Site-directed mutagenesis kits (Q5, KLD) | Generation of variant constructs for downstream in vitro functional studies (e.g., luciferase, enzymatic assays). |
| Cell Lines with Defined Genotypes | ATCC, Coriell Institute | Isogenic or disease-relevant cell models for comparing variant vs. wild-type functional impacts. |
| Protein Structure Prediction Tools | AlphaFold DB, PyMOL | Visualizing variant location to infer potential impact on protein function for PM1 (hotspot/domain) application. |
| Variant Curation Platforms | ClinGen VCI, Franklin by Genoox | Software platforms that guide and document the application of ACMG/AMP rules with current specifications. |
The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) variant classification framework provides a systematic, evidence-based methodology for interpreting genomic variants. This framework is central to modern precision medicine, translating raw genomic data into clinically actionable classifications (Pathogenic, Likely Pathogenic, Uncertain Significance, Likely Benign, Benign). This document outlines the core philosophical principles and provides detailed application notes and experimental protocols for implementing these criteria in a research setting, particularly for drug development and therapeutic target validation.
Foundational Principles & Quantitative Evidence Strengths
Table 1: Core Evidence Categories and Strength Metrics
| Evidence Category | Code | Typical Strength Weight | Key Quantitative Thresholds |
|---|---|---|---|
| Population Data | PVS1, PM2, BA1 | Very Strong (PVS1) to Standalone (BA1) | Allele frequency < 0.001% (PM2); >5% in general population (BA1) |
| Computational & Predictive Data | PP3, BP4 | Supporting (PP3/BP4) | Multiple in silico tools concur (>70% prediction for PP3; benign for BP4) |
| Functional Data | PS3, BS3 | Strong (PS3) or Supporting (BS3) | ≥80% loss of function for PS3; ≥80% wild-type function for BS3 |
| Segregation Data | PP1 | Supporting to Strong | LOD score > 2.0 (Strong); > 1.5 (Supporting) |
| De Novo Data | PS2, PM6 | Strong (PS2) to Moderate (PM6) | Confirmed paternity/maternity; ≥2 independent events for PM6 |
| Allelic Data | PM3 | Supporting to Strong | Observed in trans with pathogenic variant for recessive (Strong) |
| Hotspot & Database | PM1, PP5 | Moderate (PM1) to Supporting (PP5) | Located in critical functional domain (PM1) |
Table 2: Pathogenicity Classification Combinations
| Final Classification | Required Evidence Combination |
|---|---|
| Pathogenic (P) | 1 Very Strong (PVS1) + ≥1 Strong (PS) OR 2 Strong (PS) OR 1 Strong (PS) + ≥2 Moderate (PM) OR 1 Strong (PS) + 1 Moderate (PM) + ≥2 Supporting (PP) |
| Likely Pathogenic (LP) | PVS1 + 1 Moderate (PM) OR 1 Strong (PS) + 1-2 Moderate (PM) OR 1 Strong (PS) + ≥2 Supporting (PP) OR ≥2 Moderate (PM) |
| Uncertain Significance (VUS) | Default classification when criteria for P, LP, LB, or B are not met. |
| Likely Benign (LB) | 1 Strong (BS) OR ≥2 Supporting (BP) |
| Benign (B) | 1 Standalone (BA) OR 2 Strong (BS) |
Application Notes & Experimental Protocols
Protocol 1: Functional Assay Validation for PS3/BS3 Evidence
Objective: To quantitatively assess the impact of a genetic variant on protein function to provide Strong (PS3) or Supporting (BS3) evidence. Workflow: See Diagram 1. Detailed Methodology:
- Construct Generation: Site-directed mutagenesis is performed on a wild-type cDNA expression vector to introduce the variant of interest. All constructs are sequence-verified.
- Cell Transfection: Use an appropriate cell line (e.g., HEK293T, HeLa) deficient for the endogenous protein if possible. Transfect in triplicate with equimolar amounts of wild-type (WT), variant (VAR), and empty vector (EV) control plasmids using a standardized method (e.g., lipid-based transfection).
- Protein Harvest & Quantification: Harvest cells 48 hours post-transfection. Lyse cells in RIPA buffer with protease inhibitors. Determine total protein concentration via BCA assay. Normalize lysate concentrations.
- Functional Readout: Perform a standardized enzymatic assay, protein-protein interaction assay (e.g., co-immunoprecipitation followed by western blot), or transcriptional reporter assay specific to the protein's known function.
- Data Analysis: Normalize variant activity to WT control (set at 100%). Calculate mean and standard deviation from ≥3 independent experiments. Classification:
- PS3 Support: Statistically significant loss-of-function (≤20% of WT activity, p < 0.01).
- BS3 Support: Function not statistically different from WT (≥80% of WT activity, p > 0.05).
- Inconclusive: Activity between 20-80% of WT requires additional evidence.
Protocol 2:In SilicoAnalysis for PP3/BP4 Evidence
Objective: To aggregate computational predictions for missense variants. Methodology:
- Tool Selection: Run variant through a minimum of 5 reputable in silico predictors, encompassing different algorithms (e.g., SIFT, PolyPhen-2, REVEL, CADD, MutationTaster2025).
- Data Aggregation: Record raw scores and categorical predictions (Deleterious/Tolerated, etc.).
- Evidence Strength Assignment:
- PP3 (Supporting): ≥70% of tools predict a deleterious effect.
- BP4 (Supporting): ≥70% of tools predict a benign effect.
- Neutral/Conflicting: Results do not meet the 70% threshold for either category. Do not apply PP3 or BP4.
Protocol 3: Co-segregation Analysis for PP1 Evidence
Objective: To assess whether a variant segregates with disease phenotype in a family. Methodology:
- Pedigree & Sample Collection: Document a multi-generation pedigree. Obtain informed consent and genomic DNA from affected and unaffected family members.
- Variant Genotyping: Use targeted sequencing or PCR-based genotyping to determine variant status in all available family members.
- LOD Score Calculation: Calculate a LOD (Logarithm of Odds) score under an assumed genetic model (autosomal dominant/recessive) and penetrance.
- PP1 (Supporting): LOD score > 1.5.
- PP1 (Strong): LOD score > 2.0 with no phenocopies or non-penetrance.
- Caveats: Account for age-dependent penetrance and possibility of phenocopies.
Visualizations
Diagram 1: Functional Assay Validation Workflow
Diagram 2: ACMG/AMP Evidence Combination Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Variant Classification Research
| Item / Reagent | Function in Protocol | Example/Note |
|---|---|---|
| Site-Directed Mutagenesis Kit | Introduces specific nucleotide changes into cDNA expression vectors. | Q5 Site-Directed Mutagenesis Kit (NEB), QuikChange II. |
| cDNA Expression Vector | Backbone for expressing wild-type and variant proteins in cells. | pcDNA3.1, pCMV, or lentiviral vectors with selectable markers. |
| Competent Cells | For plasmid amplification and mutagenesis reaction transformation. | NEB 5-alpha, DH5α, Stbl3 for stable sequences. |
| Cell Line | Cellular system for functional protein expression and assay. | HEK293T (high transfection), relevant disease cell models. |
| Transfection Reagent | Delivers plasmid DNA into mammalian cells. | Lipofectamine 3000, polyethylenimine (PEI), electroporation. |
| Lysis Buffer (RIPA) | Extracts total protein from transfected cells while maintaining function. | Includes protease/phosphatase inhibitors. |
| BCA Protein Assay Kit | Quantifies total protein concentration for lysate normalization. | Essential for equal loading in functional assays. |
| Antibodies (Tag/Specific) | Detect expressed protein (via tag) or endogenous interactors. | Anti-FLAG, HA, Myc for tagged proteins; validated primary antibodies. |
| Functional Assay Substrate/Kit | Measures specific biochemical activity of the protein of interest. | Luciferase reporter, kinase activity, protein-protein binding kits. |
| Sanger Sequencing Service | Confirms variant identity in plasmids and genotyped samples. | Critical for quality control at multiple steps. |
| In Silico Prediction Tool Suite | Aggregates computational data for PP3/BP4 evidence. | REVEL, CADD, SIFT, PolyPhen-2, MutationTaster2025. |
1. Introduction: ACMG/AMP Criteria in Genomic Research & Drug Development
The 2015 consensus guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) established a standardized, evidence-based framework for classifying sequence variants. This five-tier system—Pathogenic (P), Likely Pathogenic (LP), Variant of Uncertain Significance (VUS), Likely Benign (LB), and Benign (B)—is foundational for clinical diagnostics, translational research, and therapeutic development. For drug developers, accurate classification directly impacts patient stratification for clinical trials, identification of therapeutic targets, and assessment of off-target effects. This document provides application notes and protocols for implementing these criteria within a research context.
2. Quantitative Summary of Evidence Criteria
The ACMG/AMP framework combines 28 evidence criteria, each weighted as Very Strong (VS), Strong (S), Moderate (M), or Supporting (P). Pathogenicity is assessed by combining benign and pathogenic evidence.
Table 1: ACMG/AMP Evidence Criteria Summary
| Evidence Type | Code | Weight | Example |
|---|---|---|---|
| Pathogenic Very Strong | PVS1 | VS | Null variant in a gene where LOF is a known mechanism of disease. |
| Pathogenic Strong | PS1-S4 | S | Same amino acid change as a known pathogenic variant. |
| Pathogenic Moderate | PM1-PM6 | M | Located in a mutational hot spot or well-established functional domain. |
| Pathogenic Supporting | PP1-PP5 | P | Co-segregation with disease in multiple affected family members. |
| Benign Standalone | BA1 | S | Allele frequency is >5% in population databases. |
| Benign Strong | BS1-BS4 | S | Allele frequency is greater than expected for disorder. |
| Benign Supporting | BP1-BP7 | P | Observed in trans with a pathogenic variant for a recessive disorder. |
Table 2: Rule Combinations for Final Classification
| Final Classification | Required Evidence Combination |
|---|---|
| Pathogenic (P) | 1 PVS1 + 1 PS1-PS4 OR ≥2 PS1-PS4 OR 1 PS1-PS4 + ≥3 PM1-PM6 OR 1 PS1-PS4 + 2 PM1-PM6 + ≥2 PP1-PP5 |
| Likely Pathogenic (LP) | 1 PVS1 + 1 PM1-PM6 OR 1 PS1-PS4 + 1-2 PM1-PM6 OR ≥3 PM1-PM6 OR ≥2 PM1-PM6 + ≥2 PP1-PP5 |
| Variant of Uncertain Significance (VUS) | Evidence criteria for neither Benign nor Pathogenic are met. |
| Likely Benign (LB) | 1 BS1-BS4 + 1 BP1-BP7 OR ≥2 BP1-BP7 |
| Benign (B) | 1 BA1 OR ≥2 BS1-BS4 |
3. Experimental Protocols for Evidence Generation
Protocol 3.1: In Silico and Population Frequency Analysis (Supporting Evidence: PP3/BP4, BS1/BA1)
- Objective: Assess variant frequency and computational predictions of pathogenicity.
- Methodology:
- Database Query: Interrogate population genomics databases (gnomAD, 1000 Genomes) for allele frequency. An allele frequency significantly higher than the disease prevalence is evidence for benignity (BS1).
- Computational Tool Suite: Run the variant through a curated set of in silico prediction tools.
- For Missense Variants: Use REVEL, MetaLR, SIFT, PolyPhen-2.
- For Splice Variants: Use SpliceAI, MaxEntScan, NNSPLICE.
- Evidence Assignment: Apply PP3 if multiple tools concordantly predict deleteriousness. Apply BP4 if multiple tools concordantly predict benignity. Concordance is typically defined as ≥70% of tools agreeing.
Protocol 3.2: Functional Assay for Missense Variants (Moderate Evidence: PS3/BS3)
- Objective: Empirically determine the functional impact of a variant on protein activity.
- Methodology (Example: Luciferase Reporter Assay for Transcriptional Activator):
- Construct Generation: Clone the cDNA of the wild-type (WT) and variant (VAR) gene into an appropriate mammalian expression vector.
- Cell Culture & Transfection: Seed HEK293T cells in a 96-well plate. Co-transfect cells with:
- WT or VAR expression plasmid.
- A luciferase reporter plasmid containing the gene's DNA-binding site.
- A Renilla luciferase control plasmid for normalization.
- Assay & Measurement: 48 hours post-transfection, lyse cells and measure Firefly and Renilla luciferase activity using a dual-luciferase assay kit.
- Data Analysis: Normalize Firefly luminescence to Renilla. Set WT activity to 100%. Calculate VAR activity as a percentage of WT. Evidence Assignment: PS3 if activity is <20% of WT (severe loss-of-function). BS3 if activity is >80% of WT (normal function). Results between 20-80% may be inconclusive or provide supporting evidence.
Protocol 3.3: Segregation Analysis (Supporting/Strong Evidence: PP1/PS4)
- Objective: Determine if the variant co-segregates with disease phenotype within a family.
- Methodology:
- Pedigree Construction & Sample Collection: Construct a detailed pedigree. Obtain informed consent and genomic DNA from multiple affected and unaffected family members.
- Variant Genotyping: Perform targeted sequencing (Sanger or NGS panel) for the specific variant in all collected samples.
- Statistical Analysis: Calculate a LOD (Logarithm of Odds) score under a defined genetic model (e.g., autosomal dominant). Evidence Assignment: PP1 for co-segregation in a limited number of meioses. PS4 if the segregation data achieves statistical significance (e.g., LOD score >2.0).
4. Visualizing the Classification Workflow & Biological Impact
Diagram 1: ACMG/AMP Variant Classification Workflow
Diagram 2: Biological Consequence of Variant Tiers
5. The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Variant Classification Research
| Item | Function & Application |
|---|---|
| Reference Genomic DNA (e.g., NA12878) | Standardized control for assay calibration and sequencing run QC. |
| Site-Directed Mutagenesis Kit (e.g., Q5) | For rapid generation of variant expression constructs from WT cDNA clones. |
| Mammalian Expression Vectors (e.g., pcDNA3.1) | Backbone for transient expression of WT and variant proteins in functional assays. |
| Dual-Luciferase Reporter Assay System | Gold-standard for quantifying transcriptional activity changes (PS3/BS3 evidence). |
| CRISPR-Cas9 Editing Tools (RNPs) | For creating isogenic cell lines with endogenous variant knock-in for phenotypic studies. |
| Sanger Sequencing Reagents | Orthogonal validation of NGS variants and segregation analysis in families. |
| Population Database Subscriptions (gnomAD) | Critical source for allele frequency data (BA1/BS1 evidence). |
| Variant Interpretation Platforms (e.g., Varsome, InterVar) | Bioinformatics tools to semi-automate ACMG/AMP rule application and documentation. |
The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) variant classification guidelines provide a standardized framework for interpreting the pathogenicity of genetic variants. This framework is central to clinical diagnostics, research validation, and drug target identification. The 28 criteria are stratified into categories: Pathogenic Very Strong (PVS1), Pathogenic Strong (PS1-PS4), Pathogenic Moderate (PM1-PM6), Pathogenic Supporting (PP1-PP5), Benign Standalone (BA1), Benign Strong (BS1-BS4), and Benign Supporting (BP1-BP7). Their precise application requires integration of population data, computational predictions, functional data, and segregation evidence.
| Criterion | Description | Key Quantitative Thresholds (Current Data) |
|---|---|---|
| PVS1 | Null variant in a gene where LOF is a known mechanism of disease. | Premature stop, frameshift, canonical splice site ±1/2, initiation codon, single/multi-exon deletion in gene with established LOF disease mechanism. |
| PS1 | Same amino acid change as a previously established pathogenic variant. | Must be established pathogenic variant at same residue, irrespective of nucleotide change. |
| PS2 | De novo in a patient with disease and no family history. | Confirmed paternity/maternity. For dominant disorders, ≥2 independent occurrences often required for PS2_Strong. |
| PS3 | Well-established functional studies supportive of damaging effect. | Studies in validated model systems showing severe impact on protein function/gene expression. |
| PS4 | Prevalence in affecteds significantly increased over controls. | Odds Ratio (OR) > 5.0 (p < 0.05) often considered strong; case-control studies with significant enrichment. |
| PM1 | Located in a mutational hot spot/critical functional domain. | Domain critical for function (e.g., active site of enzyme, DNA-binding domain of transcription factor). |
| PM2 | Absent from population databases (or at very low frequency). | gnomAD allele frequency < 0.0005 (or gene-specific threshold); BA1 overrides. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant. | Confirmed in trans phase (e.g., via parental testing or haplotype analysis). |
| PM4 | Protein length-changing variant (non-repeat regions). | In-frame indels, stop loss variants in non-repetitive regions. |
| PM5 | Novel missense change at an amino acid where a different pathogenic missense change has been seen. | Different nucleotide and amino acid change, but same residue. |
| PM6 | De novo without confirmation of paternity/maternity. | Unconfirmed but assumed de novo. Often used as supporting evidence. |
| PP1 | Co-segregation with disease in multiple affected family members. | LOD score > 1.9 considered moderate; > 3.0 strong. Often used as supporting. |
| PP2 | Missense variant in a gene with low rate of benign missense variation. | Gene-specific missense constraint (e.g., high Z-score in gnomAD). |
| PP3 | Multiple lines of computational evidence support a deleterious effect. | Concordant predictions from REVEL, CADD, SIFT, PolyPhen-2. REVEL > 0.75 often supportive. |
| PP4 | Patient’s phenotype highly specific for gene. | Single-gene disorder with characteristic, well-defined phenotype. |
| PP5 | Reputable source reports variant as pathogenic but evidence unavailable. | Use is discouraged in current guidelines; requires independent assessment. |
| Criterion | Description | Key Quantitative Thresholds (Current Data) |
|---|---|---|
| BA1 | Allele frequency in population databases is too high for disorder. | gnomAD AF > 5% for dominant; > 1% for recessive disorders (general thresholds). |
| BS1 | Allele frequency greater than expected for disorder. | AF above disease-specific threshold but below BA1 (e.g., 0.1%-5%). |
| BS2 | Observed in healthy adult individual(s) for a recessive, late-onset, or reduced penetrance disorder. | Homozygous in healthy adult for severe recessive pediatric disorder. |
| BS3 | Well-established functional studies show no damaging effect. | Reputable assays show normal function/expression. |
| BS4 | Lack of segregation in affected family members (non-segregation). | Failure to co-segregate in multiple families. |
| BP1 | Missense variant in gene where only truncating variants cause disease. | For genes with established LOF mechanism; missense variants not known to be pathogenic. |
| BP2 | Observed in trans with a pathogenic variant for a dominant disorder, or in cis for any disorder. | In trans with pathogenic variant in a dominant gene without compound heterozygosity expected. |
| BP3 | In-frame indels in repetitive regions without known function. | Variants in repeat regions (e.g., fibronectin type III repeats) without proven impact. |
| BP4 | Multiple lines of computational evidence suggest no impact. | Concordant benign predictions from reputable in silico tools. |
| BP5 | Variant found in case with an alternate molecular cause. | Another pathogenic variant fully explains phenotype. |
| BP6 | Reputable source reports variant as benign but evidence unavailable. | Use discouraged; requires independent assessment. |
| BP7 | Synonymous variant with no predicted impact on splicing. | Not at canonical splice sites, and splicing predictors (e.g., SpliceAI) show no impact. |
Experimental Protocols for Key Evidence Generation
Protocol 1: Functional Assays for PS3/BS3
Objective: To determine the impact of a variant on protein function in a controlled experimental system. Materials: See "Scientist's Toolkit" below. Methodology:
- Cloning & Site-Directed Mutagenesis: Clone the wild-type (WT) cDNA of the gene of interest into an appropriate expression vector (e.g., pcDNA3.1). Generate the variant construct using QuikChange or Gibson Assembly.
- Cell Transfection: Transfect HEK293T or other relevant cell lines with WT, variant, and empty vector (negative control) constructs using a lipid-based transfection reagent. Include a transfection marker (e.g., GFP).
- Protein Analysis:
- Western Blot: Harvest cells 48h post-transfection. Analyze lysates by SDS-PAGE and immunoblot for target protein and loading control (e.g., GAPDH). Quantify band intensity to assess stability/expression.
- Enzymatic/Activity Assay: Perform gene-specific functional assay (e.g., luciferase reporter for transcription factors, substrate conversion assay for enzymes). Normalize activity to protein expression or cell count.
- Localization Studies (if applicable): Perform immunofluorescence microscopy on transfected cells. Co-stain with organelle markers to assess mislocalization.
- Data Analysis: Perform ≥3 independent experiments. Compare variant to WT using Student's t-test. A significant reduction (<30% of WT activity) supports PS3. Activity comparable to WT (>80%) supports BS3.
Protocol 2: Segregation Analysis for PP1/BS4
Objective: To determine if a variant co-segregates with disease phenotype in a family. Methodology:
- Sample Collection: Obtain informed consent and collect DNA from proband and available family members (affected and unaffected).
- Genotyping: Perform Sanger sequencing or targeted NGS for the variant of interest in all family members.
- Haplotype/Phase Determination: If necessary, perform phasing via parental genotyping or long-read sequencing to determine if variants are in cis or trans.
- Statistical Evaluation: Calculate a LOD (Logarithm of Odds) score under a specified genetic model (autosomal dominant/recessive, penetrance). Use software like Superlink or Mendel. LOD score > 1.9 provides supporting (PP1) evidence; lack of segregation in a large family can support BS4.
Protocol 3:De NovoAnalysis for PS2/PM6
Objective: To confirm a variant has arisen de novo in the proband. Methodology:
- Trio Sequencing: Perform whole-exome/genome sequencing on proband and both biological parents.
- Variant Calling & Filtering: Use joint calling pipeline (e.g., GATK) to identify variants present in the proband but absent in both parents.
- Validation: Confirm candidate de novo variants by orthogonal method (Sanger sequencing) in the trio.
- Contamination Check: Use SNP arrays or sequencing data to confirm biological relationships and rule out sample swaps or contamination. Software tools: Peddy, VerifyBamID.
- Reporting: Confirmed de novo status with relationship validation supports PS2. Claimed de novo without parental confirmation is weaker evidence (PM6).
Visualizations
Decision Flow for ACMG/AMP Criteria Integration
De Novo Analysis Workflow for PS2/PM6
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in ACMG Criteria Research |
|---|---|
| gnomAD Browser | Primary resource for population allele frequency data, critical for applying BA1, BS1, PM2. |
| REVEL & CADD Scores | Meta-prediction tools aggregating multiple computational lines of evidence for PP3/BP4. |
| SpliceAI | Deep learning model to predict impact on splicing, essential for intronic/interpreting BP7. |
| Site-Directed Mutagenesis Kit | To introduce specific variants into expression constructs for functional assays (PS3/BS3). |
| HEK293T Cell Line | Common, easily transfectable mammalian cell line for in vitro functional protein studies. |
| Dual-Luciferase Reporter Assay | System to measure transcriptional activity of variants in regulatory elements or transcription factors. |
| Sanger Sequencing Reagents | Gold standard for orthogonal validation of NGS findings and segregation analysis in families. |
| Long-Read Sequencer (PacBio/ONT) | To determine haplotype phase (cis/trans) for PM3/BP2 and resolve complex variants. |
| Peddy Software | Tool to verify familial relationships and check contamination in trio sequencing for PS2. |
| LOVD / ClinVar Public Database | Curated repositories of variant classifications and evidence used for PS1, PM5, PP5/BP6. |
Introduction Within the ACMG/AMP variant classification framework research, standardization is not a theoretical ideal but an operational necessity. Inconsistencies in variant interpretation directly impact patient care, clinical trial eligibility, and drug development pipelines. This document outlines application notes and protocols to address standardization challenges, providing actionable methodologies for key stakeholders: clinical testing labs, biopharmaceutical companies, and large-scale research consortia.
Application Note 1: Inter-Laboratory Concordance Assessment
Objective: To quantify and improve concordance in variant pathogenicity classification across different diagnostic laboratories using ACMG/AMP criteria.
Key Quantitative Data Summary:
Table 1: Summary of Published Inter-Laboratory Concordance Studies (2018-2024)
| Study (Year) | Genes/Variants Assessed | Initial Concordance Rate | Major Discrepancy Rate (Pathogenic vs. Benign) | Primary Source of Discordance |
|---|---|---|---|---|
| ClinGen BRCA1/2 (2019) | 15 BRCA1/2 variants | 73% | 20% | Differing interpretation of PS3 (functional assay) and PM2 (population data) criteria. |
| ClinGen PTEN (2021) | 12 PTEN variants | 83% | 8% | Application of PP1 (co-segregation) strength and PM1 (hotspot/mutation domain) criteria. |
| CDC 2022 Pilot | 10 Variants (Multiple Genes) | 64% | 25% | Variable use of supporting (PP/BP) evidence and lack of internal calibration. |
| Recent Multi-Lab Ring Trial (2024) | 20 Challenging Variants (Oncogenics) | 91%* | 4%* | *Post-rule specification and data sharing. Residual issues with clinical validity of functional assays. |
Protocol 1.1: Structured Evidence Curation and Rule Specification
Methodology:
- Variant Selection: Assemble a panel of 10-15 historically discordant variants from public databases (ClinVar).
- Pre-Meeting Curation: Each participating laboratory independently curates all relevant evidence (population, computational, functional, segregation, de novo) into a standardized template (e.g., ClinGen Evidence Curation Interface).
- Blinded Initial Classification: Laboratories provide an initial ACMG/AMP classification without discussion.
- Structured Jury Meeting: A moderated discussion follows a fixed agenda:
- Step 1: Review and agree on the raw evidence (e.g., allele frequency, assay result).
- Step 2: Discuss and specify the rule applied to that evidence (e.g., For a functional assay: Is this a "well-established" assay? Does the result represent a "loss-of-function"? This specifies PS3/BS3 strength).
- Step 3: Re-classify based on specified rules.
- Analysis: Calculate concordance pre- and post-discussion. Document the specific rule specifications that resolved discrepancies.
Diagram 1: Inter-Lab Concordance Improvement Workflow
The Scientist's Toolkit: Reagents & Resources for Variant Curation Table 2: Essential Resources for ACMG/AMP Variant Classification
| Item | Function & Example |
|---|---|
| Standardized Curation Platform | Enforces evidence structure; enables collaboration. Example: ClinGen Evidence Curation Interface (ECI). |
| Population Frequency Databases | Provides data for BA1/BS1/PM2 criteria. Examples: gnomAD, 1000 Genomes, dbSNP. |
| In Silico Prediction Tools Suite | Provides computational evidence for PP3/BP4 criteria. Examples: Combined annotation from REVEL, CADD, SIFT, PolyPhen-2. |
| Functional Assay Standards | Validated protocols for PS3/BS3 criteria. Example: ClinGen SVI's recommendations for PTEN phosphatase assays. |
| Variant Database & Sharing Portal | Central repository for classifications and evidence. Examples: ClinVar, VICC Meta-KB. |
Application Note 2: Standardized Framework for Clinical Trial Eligibility
Objective: To define a protocol for consistent application of ACMG/AMP classifications in patient eligibility screening for genotype-driven clinical trials.
Protocol 2.1: Tiered Eligibility Determination Protocol
Methodology:
- Establish a Trial-Specific Variant Interpretation Committee (VIC): Comprised of molecular geneticists, clinical trialists, and a bioethicist.
- Define Eligibility Tiers:
- Tier 1 (Actionable): Variants classified as Pathogenic (P) or Likely Pathogenic (LP) in the specified gene(s) by an approved lab.
- Tier 2 (Investigational): Variants of Uncertain Significance (VUS) with directional evidence (e.g., VUS-favoring pathogenic, or specific functional data supporting mechanism of action of the drug).
- Tier 3 (Excluded): Benign (B) or Likely Benign (LB) variants, or VUS without supporting directional evidence.
- Centralized Review: All potential enrollee variants, especially VUS, are submitted for blinded review by the VIC using a standardized evidence portfolio (Table 3).
- Decision Documentation: The final eligibility decision and the specific evidence driving the classification (e.g., "Included based on Tier 2 due to compelling functional assay data aligned with drug mechanism") are documented in the trial master file.
Table 3: Standardized Evidence Portfolio for Trial VUS Review
| Evidence Category | Required Data Fields | Trial-Specific Consideration |
|---|---|---|
| Clinical & Phenotypic | Patient HPO terms, trial-relevant phenotypes | Does the phenotype match the drug's target pathway? |
| Molecular & Functional | Functional assay report, protein interaction data | Does the assay test a function directly modulated by the drug? |
| Computational & Predictive | REVEL score, structural modeling impact | Does the variant location affect the drug binding site? |
| Preliminary Class. | Lab's summary & ACMG/AMP code applied | Was PM1 (hotspot) applied correctly for this trial's context? |
Diagram 2: Clinical Trial Eligibility Determination Pathway
Conclusion Standardization within the ACMG/AMP ecosystem is achievable through the implementation of structured protocols for evidence curation, rule specification, and stakeholder collaboration. The application notes and detailed protocols provided here offer a concrete roadmap for improving concordance in clinical diagnostics and ensuring rigor and fairness in pharmaceutical development, ultimately accelerating the delivery of precision medicine.
From Theory to Practice: A Step-by-Step Guide to Applying ACMG-AMP Criteria
Application Notes
This protocol outlines a structured workflow for the aggregation and synthesis of evidence from disparate sources to classify genetic variants according to the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) criteria. It is designed for integration into a comprehensive research thesis focused on refining and applying these criteria. The framework systematically integrates population genomics, in silico predictions, and functional genomic assays to populate evidentiary criteria such as PM2, PP3, BS1, PS3, and others.
Core Application
The primary application is the high-throughput classification of variants of uncertain significance (VUS) in clinical and research settings, particularly for drug target validation and patient stratification in clinical trials. This structured approach minimizes classification ambiguity and supports reproducible, evidence-based decisions.
Detailed Protocols
Protocol 1: Population Frequency Data Curation for BA1/BS1/PM2 Evidence
Objective: To collect and analyze population allele frequency data to apply criteria BA1 (Benign, Stand-Alone), BS1 (Benign, Strong), or PM2 (Pathogenic, Moderate) based on absence or prevalence in reference populations.
Materials:
- Computing workstation with internet access.
- Genomic Analysis Tools: gnomAD (v4.0+), dbSNP, Bravo TopMed, or 1000 Genomes Project API/offline databases.
- Software: R (v4.3+) with packages
tidyverse,variantannotation, or Python withpandas,requests.
Methodology:
- Variant List Input: Compile a list of target variants in standard format (e.g., GRCh38, CHROM:POS:REF:ALT).
- Data Extraction (Automated Query): Use scripting to query population databases via public APIs or downloaded VCFs.
- Example gnomAD API call for variant 1-55516888-G-A:
https://gnomad.broadinstitute.org/api/?query=variant(variantId:"1-55516888-G-A", genomeBuild:GRCh38)
- Example gnomAD API call for variant 1-55516888-G-A:
- Frequency Filtering and Categorization:
- Apply ACMG-recommended frequency thresholds (e.g., BA1: >5% allele frequency in any population; BS1: frequency too high for disease severity).
- For PM2: Identify variants absent or with extremely low frequency (<0.0001) in population databases, excluding sub-populations where the variant might be common due to founder effects.
- Evidence Assignment: Populate the following table with extracted data and assign preliminary evidence codes.
Table 1: Population Frequency Analysis for Variant Classification
| Variant (GRCh38) | gnomAD v4.0 AF (Global) | gnomAD v4.0 Hom. Count | TopMed AF | Max Sub-Pop AF | Assigned ACMG Code | Justification |
|---|---|---|---|---|---|---|
| 1-55516888-G-A | 0.000032 | 0 | 0.000041 | 0.00012 (SAS) | PM2 | Absent from controls; AF << disease prevalence. |
| 2-215632451-A-G | 0.251 | 14567 | 0.243 | 0.28 (EUR) | BA1 | AF > 5%, stand-alone benign evidence. |
| 7-117199563-T-C | 0.0047 | 12 | 0.0051 | 0.011 (ASJ) | BS1 | AF significantly exceeds expected for severe childhood disorder. |
Protocol 2: ComputationalIn SilicoAnalysis for PP3/BP4 Evidence
Objective: To perform and aggregate multiple computational prediction scores to support PP3 (Pathogenic, Supporting) or BP4 (Benign, Supporting) evidence.
Materials:
- Variant effect predictor tools: Ensembl VEP (v109+), snpEff (v5.2).
- Prediction suites: REVEL, CADD, PolyPhen-2, SIFT, MutationTaster, PrimateAI, SpliceAI.
- Environment: Command-line Linux server or web-based interfaces (e.g., VEP web tool).
Methodology:
- Variant Annotation: Input the variant list into Ensembl VEP with the
--pluginflags for CADD, SpliceAI, and REVEL, or use standalone tools. - Score Aggregation: Extract pathogenicity predictions and conservation scores (e.g., GERP++).
- Consensus Rule Application: Apply pre-defined, calibrated thresholds for evidence assignment.
- PP3: Requires concordant pathogenic predictions from ≥3/5 tools (e.g., REVEL > 0.75, CADD > 25, SIFT deleterious, PolyPhen probably damaging, SpliceAI delta score > 0.2).
- BP4: Requires concordant benign predictions from ≥3/5 tools (e.g., REVEL < 0.15, CADD < 15, SIFT tolerated, PolyPhen benign).
- Evidence Logging: Record scores and final code.
Table 2: In Silico Prediction Aggregation for Variant RS12345
| Variant | REVEL | CADD | SIFT | PolyPhen-2 | SpliceAI | Consensus | ACMG Code |
|---|---|---|---|---|---|---|---|
| 1-55516888-G-A | 0.92 | 32.5 | Deleterious (0.01) | Probably Damaging (1.0) | 0.01 (No impact) | 4/5 Pathogenic | PP3 |
| 2-215632451-A-G | 0.10 | 8.2 | Tolerated (0.45) | Benign (0.12) | 0.00 | 5/5 Benign | BP4 |
Protocol 3:In VitroFunctional Assay for PS3/BS3 Evidence
Objective: To conduct a well-established functional study (e.g., luciferase reporter assay for transcriptional activity) to generate experimental evidence for PS3 (Pathogenic, Strong) or BS3 (Benign, Strong).
Materials:
- Cell Line: HEK293T or relevant cell model (ATCC).
- Plasmids: Wild-type and mutant gene-of-interest cDNA cloned into mammalian expression vector (e.g., pcDNA3.1); Reporter plasmid with responsive elements; Control Renilla luciferase plasmid (e.g., pRL-TK).
- Reagents: Transfection reagent (Lipofectamine 3000), Dual-Luciferase Reporter Assay System (Promega), cell culture media and supplements.
- Equipment: Microplate luminometer, cell culture incubator, biosafety cabinet.
Methodology:
- Cell Seeding: Seed 5 x 10^4 HEK293T cells per well in a 24-well plate 24 hours prior to transfection.
- Plasmid Transfection: Transfect each well with:
- 100 ng of wild-type or mutant expression plasmid (or empty vector control).
- 100 ng of firefly luciferase reporter plasmid.
- 10 ng of Renilla luciferase control plasmid (pRL-TK).
- Use Lipofectamine 3000 per manufacturer's protocol.
- Assay and Measurement: At 48 hours post-transfection, lyse cells and measure firefly and Renilla luciferase activity using the Dual-Luciferase Reporter Assay System on a luminometer.
- Data Analysis: Normalize firefly luciferase activity to Renilla activity for each well. Calculate mean and standard deviation from ≥3 biological replicates, each with 3 technical replicates.
- Evidence Assignment:
- PS3: Mutant shows statistically significant (p < 0.01) loss-of-function (<30% of wild-type activity) or dominant-negative effect.
- BS3: Mutant function is not statistically different from wild-type (80-120% of wild-type activity).
Table 3: Functional Assay Results for Transcriptional Activity
| Variant | Normalized Luciferase Activity (Mean ± SD) | % of Wild-Type | p-value (vs. WT) | ACMG Code |
|---|---|---|---|---|
| Wild-Type | 1.00 ± 0.12 | 100% | - | - |
| 1-55516888-G-A (Missense) | 0.18 ± 0.05 | 18% | 5.2e-8 | PS3 |
| 2-215632451-A-G (Synonymous) | 1.05 ± 0.15 | 105% | 0.45 | BS3 |
| Empty Vector | 0.02 ± 0.01 | 2% | 1.1e-10 | - |
Mandatory Visualizations
Workflow for integrating evidence for variant classification.
Luciferase reporter assay signaling pathway.
The Scientist's Toolkit
Table 4: Research Reagent Solutions for Variant Classification Workflow
| Item | Vendor/Resource Example | Function in Workflow |
|---|---|---|
| gnomAD Database | Broad Institute | Primary source of population allele frequencies for BA1/BS1/PM2 evidence. |
| Ensembl VEP | EMBL-EBI | Core tool for annotating variants and integrating in silico scores (PP3/BP4). |
| REVEL Score | dbNSFP / VEP plugin | Meta-predictor for missense variant pathogenicity; critical for PP3/BP4. |
| SpliceAI | Illumina / VEP plugin | Predicts impact on mRNA splicing, informing PVS1/PP3 evidence. |
| Dual-Luciferase Reporter Assay | Promega (Cat.# E1910) | Gold-standard kit for quantifying transcriptional activity in PS3/BS3 assays. |
| Lipofectamine 3000 | Thermo Fisher (Cat.# L3000015) | High-efficiency transfection reagent for delivering plasmids into mammalian cells. |
| Control Plasmid (pRL-TK) | Promega | Contains Renilla luciferase gene for normalization in reporter assays. |
| Precision gDNA Reference | Coriell Institute | Control samples with known genotypes for assay validation and calibration. |
Within the framework of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG AMP) guidelines for variant classification, a critical challenge lies in the consistent aggregation of individual evidence criteria. The standard "rule-based" system (e.g., PVS1, PM1, PP3) provides qualitative guidance but can lead to subjectivity in final classification. Recent research, as part of a broader thesis on refining variant classification, explores quantitative "point-based" systems to supplement the traditional approach. This document provides application notes and protocols for implementing and comparing these two systems for quantifying combined evidence strength, aimed at increasing standardization and reproducibility in clinical and research settings.
Core Systems: Definitions and Data
Table 1: Comparison of Rule-Based and Point-Based Evidence Systems
| Aspect | ACMG/AMP Rule-Based System | Proposed Point-Based System (Example Schema) |
|---|---|---|
| Foundation | Pre-defined combinations of categorical evidence codes. | Assignment of numerical weights to each evidence code. |
| Evidence Strength | Qualitative (Supporting, Moderate, Strong, Very Strong). | Quantitative (e.g., PP3 = +0.5, PM2 = +1.0, PM1 = +1.5, PVS1 = +4.0). |
| Combination Logic | Pre-specified rules for combining codes into Pathogenic/Likely Pathogenic (P/LP) or Benign/Likely Benign (B/LB) classifications (e.g., 1 Strong + 2 Moderate = LP). | Summation of weighted points, with thresholds for final classifications (e.g., ≥6.5 = LP; ≤-4.0 = LB). |
| Flexibility | Limited; ambiguous combinations require expert judgment. | High; allows for nuanced aggregation of mixed or novel evidence types. |
| Primary Goal | Clinical actionability with clear boundaries. | Quantitative transparency and research reproducibility. |
Table 2: Example Point Assignment and Thresholds (Research Schema)
| Evidence Code | Assigned Points (Pathogenic) | Assigned Points (Benign) | Rationale for Weight |
|---|---|---|---|
| PVS1 | +4.0 | N/A | Very strong predicted null effect. |
| PS1-PS4 | +2.0 to +3.0 | N/A | Strong experimental/functional evidence. |
| PM1-PM6 | +1.0 to +2.0 | N/A | Moderate evidence tier. |
| PP1-PP5 | +0.5 to +1.5 | N/A | Supporting evidence tier. |
| BA1 | N/A | -4.0 | Stand-alone benign. |
| BS1-BS4 | N/A | -1.0 to -3.0 | Strong benign evidence. |
| BP1-BP7 | N/A | -0.5 to -1.5 | Supporting benign evidence. |
| Classification Threshold | Likely Pathogenic: ≥6.5 | Likely Benign: ≤-4.0 | Derived from statistical modeling of rule-based outcomes. |
| Pathogenic: ≥8.0 | Benign: ≤-6.0 |
Experimental Protocols for System Comparison and Validation
Protocol 1: Calibration of Point Weights Using Known Variant Sets Objective: To derive and calibrate numerical weights for each ACMG/AMP criterion based on a gold-standard dataset. Materials: Curated dataset of variants with established classifications (e.g., from ClinVar, expert panels). Method:
- Assemble a training set of 500-1000 variants with expert-reviewed P/LP/B/LB classifications and fully annotated applicable evidence codes.
- Initialize arbitrary starting weights for each evidence code (e.g., PS=3, PM=2, PP=1, etc.).
- For each variant, sum the points of all applicable pathogenic and benign evidence codes to obtain a net score.
- Use logistic regression or machine learning (e.g., support vector machine) to optimize the weight values, minimizing the discrepancy between net score-derived classifications and the gold-standard classifications.
- Validate the optimized weights on a separate, held-out test dataset of variants.
- Establish classification thresholds (P/LP/B/LB) by analyzing the distribution of net scores for each class in the training set.
Protocol 2: Inter-Rater Concordance Study Objective: To measure the improvement in classification consistency when using a point-based system versus the rule-based system alone. Materials: Panel of at least 5 variant scientists; set of 50 complex VUS (Variants of Uncertain Significance) with rich but conflicting evidence. Method:
- Provide all participants with identical variant evidence dossiers (clinical, computational, functional data).
- Phase 1 (Rule-Based): Each participant applies standard ACMG/AMP criteria to assign evidence codes and a final classification (P/LP/VUS/LB/B) for each variant. Do not discuss.
- Collect and calculate inter-rater agreement (Fleiss' Kappa) for final classifications.
- Phase 2 (Point-Based): Provide participants with the calibrated point-weight table (from Protocol 1) and a threshold guide.
- Participants re-classify the same variants by assigning codes, summing points, and applying thresholds.
- Calculate inter-rater agreement for Phase 2 classifications.
- Analysis: Compare Kappa statistics between Phase 1 and Phase 2. A significant increase indicates improved standardization.
Visualization of Workflow and System Integration
Dual Pathway for Variant Classification
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Evidence Quantification Research
| Item / Resource | Function / Application in This Research |
|---|---|
| Curated Variant Databases (ClinVar, LOVD) | Provide gold-standard sets of classified variants with evidence annotations for system training and validation. |
| Variant Annotation Suites (VEP, ANNOVAR, InterVar) | Automate the initial gathering and scoring of computational evidence (PP3/BP4, PM2, etc.) for high-throughput analysis. |
| Statistical Software (R, Python with scikit-learn) | Essential for performing logistic regression, machine learning, and kappa statistic calculations to calibrate weights and measure concordance. |
| Consensus Classification Platform (e.g., Franklin by Genoox, VIC) | Enables blinded multi-rater studies and captures the decision-making process for both rule-based and point-based approaches. |
| Calibrated Point-Weight Reference Table | The core output of Protocol 1; serves as the key reagent for implementing the quantitative system in research or pilot clinical settings. |
| ACMG/AMP Classification Guidelines (Original & Updated) | The foundational document against which any quantitative system must be benchmarked to ensure clinical relevance. |
Within the broader research on the ACMG/AMP variant classification criteria, a critical gap exists in the explicit delineation of guidelines for somatic (cancer) versus germline variant interpretation. This article presents application notes and protocols to address this gap, focusing on the specialized, context-dependent application of evidence criteria across these two distinct genomic landscapes. The overarching thesis posits that a unified but adaptable framework is essential for accurate variant classification in precision oncology and heritable disease risk assessment.
Comparative Framework: Somatic vs. Germline Guidelines
Foundational Principles and Objectives
The primary objective in somatic variant analysis is to identify actionable alterations that drive tumorigenesis, guide therapy, or predict prognosis. In contrast, germline variant analysis aims to identify heritable pathogenic variants that confer disease risk to the proband and potentially their relatives.
Key Differences in Evidence Strength and Application
Quantitative differences in the application of ACMG/AMP criteria are summarized below.
Table 1: Comparative Application of ACMG/AMP Criteria in Somatic vs. Germline Contexts
| ACMG/AMP Criterion | Application in Germline Variants | Application in Somatic Variants (Cancer) | Rationale & Key References (ClinGen SVI, AMP/CAP/ASCO) |
|---|---|---|---|
| Population Data (PM2/BA1) | GnomAD frequency critical for rare disease. BA1 if >5% in population. | Population frequency less relevant. Focus on tumor-specific databases (e.g., COSMIC). PM2 not routinely applied. | Somatic driver mutations are often rare in general populations but recurrent in tumors. |
| Computational Evidence (PP3/BP4) | In silico predictions weighted for missense variants. | Critical for hotspot missense mutations (e.g., TP53, KRAS). Stronger PP3 for known oncogenic hotspots. | Recurrent mutations at specific residues have established oncogenic computational profiles. |
| Functional Data (PS3/BS3) | Controlled experimental models (in vitro, animal). Required for definitive classification. | May use cancer-specific functional assays (e.g., cell proliferation, transformation). Clinical response to targeted therapy can support PS3. | Functional impact is defined by oncogenic properties, not just loss/gain-of-function. |
| Variant Hotspots (PS1) | Used for known pathogenic missense changes at same codon. | Greatly strengthened. Recurrence at same amino acid in cancer cohorts is strong independent evidence (often PS1_moderate/strong). | Tumor type-specific recurrence is a hallmark of driver mutations. |
| De Novo (PS2/PM6) | Evidence for de novo occurrence in proband. | Not applicable (somatic variants are, by definition, de novo in the tumor). | Replaced by assessing variant allele frequency (VAF) and clonality within tumor. |
| Allelic Frequency (PM3) | Observation in trans with a pathogenic variant for recessive disorders. | Observation in specific cis/trans configurations with other somatic variants (e.g., compound heterozygous hits in TSGs) can be supportive. | Context of co-occurring mutations defines oncogenic pathways. |
| Patient Phenotype (PP4) | Match with specific genetic disorder. | Match with tumor type/histology and biomarker profile associated with the variant (e.g., BRCA2 in ovarian CA). | Must align with disease-specific molecular signatures. |
| Reputable Source (PP5) | Use with caution; not standalone. | Use with extreme caution. Curation in somatic databases (OncoKB, CIViC) may carry stronger weight but requires independent review. | Somatic knowledge bases are clinically oriented but dynamic. |
Detailed Experimental Protocols
Protocol: Functional Validation of a Somatic Missense Variant via Cell Proliferation and Colony Formation Assay
Objective: To provide experimental evidence (PS3/BS3) for the oncogenic potential of a somatic missense variant in a putative oncogene.
Materials:
- Isogenic cell line pair (e.g., NIH/3T3, MCF10A) engineered to express wild-type (WT) or mutant (MUT) allele of the gene of interest.
- Complete growth medium (appropriate for cell line).
- Dulbecco’s Phosphate Buffered Saline (DPBS).
- 0.25% Trypsin-EDTA.
- Hemocytometer or automated cell counter.
- 6-well and 96-well tissue culture plates.
- Crystal violet stain (1% w/v in 20% methanol) or Cell Counting Kit-8 (CCK-8).
- Software for statistical analysis (e.g., GraphPad Prism).
Methodology:
- Cell Seeding for Proliferation:
- Harvest and count WT and MUT cells.
- Seed 1,000-2,000 cells per well in a 96-well plate (6-8 replicates per line).
- Incubate at 37°C, 5% CO₂.
Proliferation Measurement (Days 1-7):
- Option A (CCK-8): At 24h intervals, add 10 µL CCK-8 reagent to designated wells. Incubate for 2-4 hours. Measure absorbance at 450 nm using a plate reader.
- Option B (Crystal Violet): At desired time points, fix cells in designated wells with 4% PFA for 20 min, stain with crystal violet for 15 min, wash, solubilize in 10% acetic acid, and measure absorbance at 590 nm.
Cell Seeding for Colony Formation:
- Seed 500-1000 cells per well in a 6-well plate (in triplicate).
- Incubate for 10-14 days, refreshing medium every 3-4 days.
Colony Staining and Quantification:
- Aspirate medium, wash with PBS, fix with 4% PFA for 20 min.
- Stain with crystal violet for 15 min. Rinse gently with water and air-dry.
- Image plates and count colonies (>50 cells) manually or using software (e.g., ImageJ).
Data Analysis:
- Plot proliferation curves (Mean ± SD absorbance vs. Time). Perform statistical comparison (e.g., two-way ANOVA).
- Calculate colony forming efficiency: (Number of colonies / Number of cells seeded) * 100%. Compare WT vs. MUT using Student's t-test.
- Interpretation: A statistically significant increase in proliferation rate and/or colony formation for MUT cells supports oncogenic gain-of-function (PS3). No difference supports a benign finding (BS3).
Protocol: Computational Oncogenicity Assessment for Somatic Missense Variants
Objective: To apply and integrate in silico predictions (PP3/BP4) tailored for cancer.
Materials:
- Variant list (e.g., VCF file).
- Access to computational tools:
- Cancer-specific: CHASM, CanPredict, TransFIC.
- General/Pan-cancer: REVEL, MetaSVM, CADD, SIFT, PolyPhen-2.
- Access to hotspot databases: MSK-IMPACT Hotspots, cBioPortal.
Methodology:
- Data Input: Format variant data (Chromosome, Position, Ref, Alt, Gene).
- Parallel Analysis:
- Step A - General Predictors: Run variant through ensemble predictors (REVEL, CADD). Use established cutoffs (e.g., REVEL > 0.75 suggests pathogenic).
- Step B - Cancer-Specific Predictors: Submit variant to CHASM/CanPredict for tumor type-specific oncogenicity scores.
- Step C - Hotspot Check: Query COSMIC, cBioPortal for recurrence of the exact amino acid change in the same cancer type.
- Evidence Integration:
- Strong PP3: Variant is a known hotspot (Step C) AND has supportive scores from both general and cancer-specific predictors.
- Moderate PP3: Variant is not a known hotspot but has concordantly pathogenic scores from >2 tools, including one cancer-specific tool.
- BP4: Multiple (>3) reputable computational tools suggest a benign impact. Cancer-specific tools are non-predictive of oncogenicity.
- Documentation: Record all scores, databases queried, and final integrated judgment.
Visualization: Pathways and Workflows
Title: Somatic vs Germline Variant Analysis Workflow
Title: Differential Weight of ACMG Evidence Criteria
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Somatic Variant Functional Characterization
| Item | Function & Application in Cancer Research | Example Product/Catalog |
|---|---|---|
| Isogenic Cell Line Pairs | Gold-standard for comparing variant effect; engineered via CRISPR-Cas9 to contain WT vs. mutant allele in same genetic background. | Horizon Discovery (e.g., HAP1 isogenic lines), ATCC engineered lines. |
| Cancer-Specific Functional Assay Kits | Quantify oncogenic phenotypes: proliferation, invasion, colony formation. | Cell Counting Kit-8 (CCK-8, Dojindo), CellTiter-Glo (Promega), Cultrex Cell Invasion Assay (Bio-Techne). |
| Phospho-Specific Antibodies | Detect activation of signaling pathways downstream of oncogenic variants (e.g., p-ERK, p-AKT). | CST (Cell Signaling Technology) Phospho-AKT (Ser473) #4060. |
| Ba/F3 Proliferation Assay System | IL-3-dependent murine pro-B cell line used to test oncogenic transformation by conferring cytokine-independent growth. | DSMZ (ACC 300), routinely engineered with gene variants. |
| Oncogenic Pathway Reporter Kits | Luciferase-based reporters for pathways commonly altered in cancer (e.g., TGF-β, Wnt, NF-κB). | Cignal Reporter Assay Kits (Qiagen). |
| Targeted Therapy Inhibitors | Used in functional rescue experiments to demonstrate variant-specific drug sensitivity (PS3 support). | Selleckchem chemical inhibitors (e.g., Vemurafenib for BRAF V600E). |
| High-Fidelity DNA Polymerase | Critical for error-free amplification of templates for site-directed mutagenesis to create mutant constructs. | Q5 Hot-Start High-Fidelity 2X Master Mix (NEB). |
| Next-Generation Sequencing Library Prep Kits | For targeted sequencing to confirm engineered mutations and rule off-target effects in cellular models. | Illumina TruSeq Custom Amplicon, Twist NGS Panels. |
Application Notes
This document details protocols for integrating Next-Generation Sequencing (NGS) data analysis with high-throughput functional screening to classify variants according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) criteria. This integration is critical for advancing the thesis research on refining these classification frameworks, providing both computational (PS3/BS3) and functional (PS3/BS3, PM2, PP3/BP4) evidence.
Table 1: Comparison of High-Throughput Screening Platforms for Variant Functional Assessment
| Platform | Throughput (Variants/Week) | Assay Type | Key Readout | Typical Turnaround Time | Approx. Cost per Variant | Primary ACMG/AMP Evidence Generated |
|---|---|---|---|---|---|---|
| Deep Mutational Scanning (DMS) | 1,000 - 10,000 | In vitro selection + NGS | Fitness score, enrichment | 4-6 weeks | $10 - $50 | PS3, BS3, PP3 |
| Massively Parallel Reporter Assay (MPRA) | 5,000 - 50,000 | Cell-based transfection + NGS | Transcriptional activity | 3-4 weeks | $5 - $20 | PS3, BS3, PP3 (for non-coding) |
| Pooled CRISPR Screening | 10,000 - 100,000 | Cell-based knockout/activation | Cell growth, fluorescence | 5-8 weeks | $2 - $10 | PS3, BS3 (for LoF/GoF) |
| Multiplexed Assays of Variant Effect (MAVEs) | 5,000 - 20,000 | Protein stability/function | Fluorescence, binding | 4-6 weeks | $20 - $100 | PS3, BS3 |
Table 2: NGS Analysis Metrics for Variant Classification Support
| Analysis Step | Key Metric | Target Threshold for High-Confidence Call | Impact on ACMG/AMP Criteria |
|---|---|---|---|
| Sequencing | Mean Coverage Depth | ≥100x for germline; ≥500x for somatic | PM2 (Absent from controls) |
| Variant Calling | SNV Quality Score (QUAL) | ≥100 | Supports all criteria |
| Variant Filtering | Population Frequency (gnomAD) | < 0.0001 for dominant; < 0.01 for recessive | PM2, BS1, BA1 |
| In Silico Prediction | REVEL Score Pathogenicity | > 0.75 (Strong Pathogenic) < 0.15 (Benign) | PP3, BP4 |
Experimental Protocols
Protocol 1: Integrated NGS Pipeline for Variant Prioritization
Objective: To identify and prioritize rare, potentially pathogenic variants from patient cohorts for downstream functional screening. Duration: 2-3 days of compute time.
Materials (Research Reagent Solutions):
- Input: DNA/RNA samples, Target capture probes (e.g., Twist Comprehensive Exome Panel).
- Sequencing Kit: Illumina NovaSeq X Plus 10B Kit.
- Alignment Reference: GRCh38/hg38 human genome assembly.
- Variant Caller: GATK (v4.5) or DRAGEN (v4.2) pipeline.
- Annotation Tools: Ensembl VEP (v111) or ANNOVAR.
- Population Databases: gnomAD (v4.0), 1000 Genomes.
- In Silico Tools: dbNSFP (v4.3a) containing REVEL, SIFT, PolyPhen-2.
Methodology:
- Library Preparation & Sequencing: Perform exome or panel capture using a standardized kit. Sequence on an Illumina platform to achieve a minimum mean coverage of 100x.
- Primary Analysis (Base Calling): Use Illumina DRAGEN or bcftools for FASTQ generation and demultiplexing.
- Secondary Analysis (Alignment & Variant Calling):
- Align reads to GRCh38 using DRAGEN or BWA-MEM.
- Mark duplicate reads using Picard Tools.
- Perform variant calling for SNVs and INDELs using GATK HaplotypeCaller in GVCF mode, followed by joint genotyping across all samples.
- Tertiary Analysis (Annotation & Filtering):
- Annotate the VCF file with population frequencies (gnomAD), in silico predictions (REVEL, CADD), and clinical databases (ClinVar).
- Apply hard filters: Read depth (DP ≥ 10), genotype quality (GQ ≥ 20), alternate allele fraction (for heterozygotes).
- Prioritize variants based on: rarity (gnomAD allele frequency < 0.001), predicted impact (missense, splice, LoF), and combined annotation score (e.g., REVEL > 0.7).
- Output: A ranked list of Variants of Uncertain Significance (VUS) with associated computational evidence (PM2, PP3, BP4, BP7) for functional testing.
Diagram: NGS Analysis to Variant Prioritization Workflow
Protocol 2: High-Throughput Functional Validation via Deep Mutational Scanning (DMS)
Objective: To experimentally assess the functional impact of hundreds of prioritized missense VUS in a gene of interest in a single experiment, generating strong functional evidence (PS3/BS3).
Materials (Research Reagent Solutions):
- Variant Library: Saturation mutagenesis oligo pool for the target gene (e.g., from Twist Bioscience).
- Cloning System: Restriction enzyme (e.g., SapI) for Golden Gate assembly, or Gibson assembly mix.
- Expression Vector: Lentiviral or yeast display vector with a selectable reporter (fluorescence, antibiotic resistance).
- Cell Line: Appropriate mammalian (HEK293T) or yeast model system.
- Selection Reagents: Antibiotics (Puromycin), FACS sorting buffers, NGS library prep kit for post-selection analysis.
- Analysis Software: Enrich2 or DiMSum for DMS data analysis.
Methodology:
- Variant Library Construction:
- Design oligonucleotides encoding all possible single-nucleotide substitutions for the target protein domain.
- Use pooled oligo synthesis to generate the DNA library. Clone the library into the expression vector via high-efficiency Golden Gate assembly.
- Transform the plasmid library into E. coli, harvest plasmid DNA, and sequence to confirm library representation.
- Delivery & Expression:
- For mammalian systems, produce lentivirus from the plasmid library and transduce cells at a low MOI to ensure single-variant integration. Select with puromycin.
- For yeast, perform direct transformation and select on appropriate media.
- Functional Selection:
- Apply the relevant functional pressure (e.g., growth factor deprivation for a kinase, ligand binding competition, temperature stress).
- Collect the pre-selection (input) population and the post-selection (output) population after several cell doublings or rounds of selection.
- Sequencing & Enrichment Analysis:
- Isolate genomic DNA (for integrated libraries) or plasmid DNA from input and output populations.
- Amplify the variant region by PCR and perform NGS (MiSeq) to high depth (~500x per variant).
- Quantify the frequency of each variant in the input and output pools using Enrich2 software. Calculate a functional score (log2 enrichment ratio).
- Interpretation for ACMG/AMP:
- Variants with significantly decreased abundance (score < -2.0) are classified as Functional Loss, supporting PS3.
- Variants with no significant change from wild-type (score between -0.5 and 0.5) are classified as Functionally Normal, supporting BS3.
- Document all parameters for the PS3/BS3 strength calibration (replicates, effect size, statistical confidence).
Diagram: Deep Mutational Screening (DMS) Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in NGS/HTS Integration | Example Product/Kit |
|---|---|---|
| Hybridization Capture Probes | Enriches genomic regions of interest (e.g., exome, gene panel) prior to sequencing for efficient variant discovery. | Twist Comprehensive Exome Panel, IDT xGen Pan-Cancer Panel |
| Ultra-High-Fidelity PCR Mix | Amplifies target regions from pooled genomic DNA with minimal error for DMS library preparation and NGS amplicon sequencing. | Q5 High-Fidelity DNA Polymerase (NEB), KAPA HiFi HotStart ReadyMix |
| Pooled Oligo Library | Synthesizes thousands of defined variant sequences in parallel for constructing saturation mutagenesis libraries. | Twist Bioscience Custom Pooled Oligo Libraries, Agilent SureEdit |
| Lentiviral Packaging System | Produces high-titer, replication-incompetent lentivirus for efficient, stable delivery of variant libraries into mammalian cells. | psPAX2/pMD2.G packaging plasmids (Addgene), Lenti-X Packaging Single Shots (Takara) |
| Chromatin Conformation Kit | Assays 3D genome structure (e.g., Hi-C) to inform MPRA design for non-coding variants in regulatory elements. | Arima-HiC Kit, Dovetail Omni-C Kit |
| CRISPR Knockout Pooled Library | Screens for gene essentiality or variant-specific synthetic lethality in a high-throughput format. | Brunello whole-genome CRISPRko library (Broad), Custom sgRNA libraries (Synthego) |
| NGS Multiplexing Indexes | Uniquely tags samples or experimental conditions, allowing pooling and parallel sequencing. | Illumina IDT for Illumina UD Indexes, Nextera XT Index Kit |
| Variant Analysis Suite | Integrated software for secondary/tertiary NGS analysis, annotation, and ACMG classification. | DRAGEN Bio-IT Platform (Illumina), VarSome Clinical |
Application Note: These case studies demonstrate the rigorous application of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) variant classification criteria within a modern genomics research setting. The framework is integral to a broader thesis investigating the refinement and consistent application of these criteria, particularly for challenging genomic alterations encountered in translational research and therapeutic development.
Case Study 1: Classifying a Novel Missense Variant inKRAS(p.Gly12Cys)
Clinical and Genomic Context
A novel KRAS missense variant, c.34G>T (p.Gly12Cys), was identified via next-generation sequencing (NGS) in a colorectal adenocarcinoma biopsy. This variant is absent from population databases (gnomAD) and clinical archives (ClinVar).
Table 1: In Silico Prediction and Population Frequency Data for KRAS p.Gly12Cys
| Data Type | Source/Tool | Result | Interpretation for ACMG |
|---|---|---|---|
| Population Frequency | gnomAD v4.0.0 | 0/1,000,000 alleles | Supports PM2 (Absent from controls) |
| Computational Evidence | REVEL | Score: 0.92 | Supports PP3 (Pathogenic computational prediction) |
| SIFT | Deleterious (0.00) | ||
| PolyPhen-2 | Probably Damaging (1.000) | ||
| Functional Data | Published assay (McCarthy et al., 2022) | Increased GTP binding & p-ERK signaling | Supports PS3 (Functional studies supportive) |
| Variant Location | UniProt | GTPase domain, Gly12 residue | Supports PM1 (Located in mutational hot spot) |
| De Novo Observation | N/A | Not observed | N/A |
Experimental Protocol: Functional Assay for KRAS Mutant Activity
Protocol Title: Luciferase Reporter Assay for RAS/MAPK Pathway Activation
Objective: Quantify the functional impact of the KRAS p.Gly12Cys variant on downstream MAPK signaling.
Materials:
- HEK293T cells (ATCC CRL-3216)
- Expression plasmids: pcDNA3.1-KRAS (WT, p.Gly12Cys, p.Gly12Asp [positive control])
- Reporter plasmid: pGL4.30[luc2P/NFAT-RE/Hygro] or pSRE-Luc (for serum response element reporting)
- Transfection reagent (e.g., Lipofectamine 3000)
- Dual-Luciferase Reporter Assay System
- Luminometer
Methodology:
- Cell Seeding: Seed HEK293T cells in 24-well plates at 1x10^5 cells/well 24 hours prior to transfection.
- Plasmid Transfection: Co-transfect each well with 200 ng of KRAS expression plasmid, 200 ng of firefly luciferase reporter plasmid (SRE-dependent), and 20 ng of Renilla luciferase control plasmid (pRL-TK) for normalization. Include triplicates for each construct.
- Incubation: Incubate cells for 48 hours post-transfection under standard conditions (37°C, 5% CO2).
- Lysate Preparation: Lyse cells using 1X Passive Lysis Buffer. Gently shake plates for 15 minutes at room temperature.
- Luciferase Measurement: Transfer 20 µL of lysate to a white-walled plate. Initiate firefly luciferase reaction by injecting 50 µL of Luciferase Assay Reagent II, measure luminescence. Quench reaction and activate Renilla luciferase by injecting 50 µL of Stop & Glo Reagent, measure luminescence.
- Data Analysis: Calculate the ratio of firefly to Renilla luciferase activity for each well. Normalize the activity of mutant constructs to the wild-type KRAS control. Statistical significance is determined via an unpaired t-test (p < 0.05).
ACMG Classification Path (KRAS p.Gly12Cys):
- Pathogenic Moderate (PM) Criteria: PM1 (hotspot), PM2 (absent from controls), PM5 (novel missense change at established pathogenic residue).
- Pathogenic Supporting (PP) Criteria: PP3 (computational evidence).
- Pathogenic Strong (PS) Criteria: PS3 (confirmed functional impact).
- Overall Classification: Pathogenic (PS3 + PM1 + PM2 + PM5 + PP3).
Diagram 1: KRAS variant classification workflow.
Case Study 2: Classifying a Complex Structural Variant inBRCA1
Clinical and Genomic Context
A multi-exon duplication of uncertain significance was detected in BRCA1 (exons 3-9 duplication) via chromosomal microarray in a patient with early-onset breast cancer. Breakpoint analysis suggested a tandem duplication.
Table 2: Evidence for Classifying the BRCA1 Exons 3-9 Duplication
| Data Type | Method/Evidence | Result | Interpreting for ACMG |
|---|---|---|---|
| Variant Type | Microarray, LR-PCR & Sequencing | Tandem in-frame duplication of exons 3-9 | PVS1 strength? (See analysis) |
| Population Data | Internal DB, gnomAD-SV | Not observed | Supports PM2 |
| Case-Control Data | Literature mining | Similar duplications reported as pathogenic | Supports PS4 (Patient phenotype specific) |
| Segregation Data | Family testing | Co-segregates with disease in 3 affected relatives | Supports PP1_Strong |
| Gene Function | Curation | Loss-of-function mechanism known | Required for PVS1 application |
| RNA Analysis | RT-PCR from patient LCLs | Aberrant splicing, frameshift transcript | Confirms PVS1 (null effect) |
Experimental Protocol: Breakpoint Confirmation and RNA Analysis
Protocol Title: Long-Range PCR and Reverse Transcription PCR for Structural Variant Characterization
Objective: Confirm the genomic breakpoints of the BRCA1 duplication and assess its impact on mRNA.
Materials:
- Patient genomic DNA and lymphoblastoid cell line (LCL) RNA.
- High-fidelity, long-range PCR kit (e.g., PrimeSTAR GXL).
- BRCA1-specific primers flanking predicted breakpoint region.
- Reverse transcription kit (e.g., SuperScript IV).
- PCR reagents for cDNA amplification.
- Agarose gel electrophoresis and Sanger sequencing.
Methodology - Part A (Genomic Confirmation):
- Primer Design: Design outward-facing primers in putative duplicated and flanking unique sequences based on microarray data.
- Long-Range PCR: Set up 50 µL reactions per manufacturer's protocol. Use 100 ng patient gDNA. Cycle conditions: 98°C for 2 min; 35 cycles of 98°C for 10s, 68°C for 5 min; final extension at 68°C for 10 min.
- Analysis: Resolve PCR products on a 0.8% agarose gel. Purify the unique amplicon and perform Sanger sequencing to define exact breakpoints.
Methodology - Part B (RNA Impact Analysis):
- cDNA Synthesis: Convert 1 µg of total RNA from patient and control LCLs to cDNA using random hexamers and SuperScript IV.
- RT-PCR: Design primers in BRCA1 exons 2 and 10. Amplify cDNA using standard PCR.
- Product Analysis: Resolve products on a 2% agarose gel. Expect a larger product from the patient if duplication is included, or a smaller/shifted product if splicing is altered. Purify and sequence aberrant bands.
ACMG Classification Path (BRCA1 Exons 3-9 Dup):
- Pathogenic Very Strong (PVS) Criteria: PVS1 (confirmed null variant via RNA study – frameshift).
- Pathogenic Strong (PS) Criteria: PS4 (prevalence in affected individuals increased over controls).
- Pathogenic Moderate (PM) Criteria: PM2 (absent from population databases).
- Supporting (PP) Criteria: PP1_Strong (co-segregation with disease).
- Overall Classification: Pathogenic (PVS1 + PS4 + PM2 + PP1_Strong).
Diagram 2: BRCA1 structural variant analysis flow.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Variant Classification Studies
| Reagent/Material | Provider Examples | Primary Function in Classification |
|---|---|---|
| High-Fidelity DNA Polymerase (Long-Range) | Takara Bio (PrimeSTAR GXL), Thermo Fisher (Platinum SuperFi II) | Accurate amplification of large genomic fragments for SV breakpoint validation. |
| Dual-Luciferase Reporter Assay System | Promega | Quantitative measurement of transcriptional activity to assess variant impact on signaling pathways (e.g., MAPK). |
| Next-Generation Sequencing Kits (Illumina) | Illumina (Nextera Flex), Twist Bioscience | Comprehensive variant detection across all variant classes (SNVs, Indels, SVs). |
| Cell Lines (HEK293T, LCLs) | ATCC, Coriell Institute | Consistent cellular models for in vitro functional assays and RNA studies. |
| CRISPR/Cas9 Gene Editing Systems | Integrated DNA Technologies (IDT), Synthego | Isogenic cell line generation for controlled functional studies. |
| Splicing Reporter Minigenes | Custom vector services (GeneArt, GenScript) | Assessment of variant impact on mRNA splicing patterns. |
| Population Variant Databases | gnomAD, dbSNP, DECIPHER | Critical resources for evaluating variant frequency against healthy populations (ACMG criterion PM2). |
| In Silico Prediction Suites | Varsome, Franklin by Genoox, InterVar | Aggregated computational evidence for pathogenicity (PP3/BP4 criteria). |
Navigating Grey Areas: Solutions for Common Challenges in Variant Classification
Application Notes and Protocols
Within the framework of ACMG/AMP variant classification criteria research, the resolution of Variants of Uncertain Significance (VUS) remains a critical bottleneck in genomic medicine. This document outlines contemporary strategies and practical protocols for VUS characterization, integrating functional assays, computational approaches, and data sharing to enable definitive pathogenicity classification.
1. Quantitative Landscape of VUS Prevalence and Resolution Rates
Table 1: VUS Statistics Across Major Genomic Databases (as of 2024)
| Database / Study | Total Variants Analyzed | Reported VUS Rate | Average Re-classification Rate (Annual) | Primary Re-classification Driver |
|---|---|---|---|---|
| ClinVar (Aggregate) | ~2.1 million submissions | ~33% | ~4.2% | New functional data & allele frequency |
| gnomAD v4.0 | ~807,000 exomes/genomes | Not applicable (controls) | N/A | N/A |
| BRCA1/2-specific studies | ~50,000 variants | ~20-40% (historical) | ~10-15% (targeted effort) | Splicing assays & family segregation |
| Cardiomyopathy Panels | ~15,000 unique variants | ~40-50% | ~3-5% | In silico predictors & case cohorts |
2. Experimental Protocols for Functional Characterization
Protocol 2.1: High-Throughput Splicing Assay (MaPSy)
- Objective: Quantify the impact of a VUS on mRNA splicing.
- Materials: Genomic DNA containing VUS, minigene vector (e.g., pSpliceExpress), HEK293T cells, transfection reagent, RT-PCR reagents, capillary electrophoresis system (e.g., Fragment Analyzer).
- Method:
- Cloning: Amplify a genomic fragment (~500 bp) flanking the exon containing the VUS. Clone into the minigene vector upstream of a reporter exon.
- Site-Directed Mutagenesis: Generate the VUS and a known pathogenic/likely benign control.
- Transfection: Transfect wild-type, VUS, and control constructs into HEK293T cells in triplicate.
- RNA Isolation & RT-PCR: Isolve total RNA 48h post-transfection. Perform RT-PCR using vector-specific primers flanking the cloned insert.
- Analysis: Resolve PCR products by capillary electrophoresis. Quantify the percentage of transcripts with exon skipping, inclusion, or intron retention. A >20% change in splicing pattern relative to wild-type is considered significant.
Protocol 2.2: Saturation Genome Editing (SGE) for Functional Assessment
- Objective: Determine the functional consequence of all possible single-nucleotide variants in a critical exon or domain.
- Materials: HAP1 or RPE1 cells, CRISPR-Cas9 reagents, donor template library encoding all possible SNVs, next-generation sequencer, flow cytometer (if using phenotypic sort).
- Method:
- Library Design: Synthesize an oligonucleotide pool encoding all possible nucleotide changes for the target region within a homology-directed repair (HDR) template.
- Delivery & Selection: Co-electroporate cells with Cas9 ribonucleoprotein (targeting the site) and the donor library. Apply antibiotic selection for integrated donors.
- Phenotyping: Culture cells for 10-14 population doublings. Either sort cells based on a relevant phenotype (e.g., surface marker loss) or harvest genomic DNA from the population over multiple time points.
- Deep Sequencing: Amplify the edited genomic region from sorted/collected samples and the initial library. Sequence deeply (>500x coverage).
- Analysis: Calculate the enrichment or depletion of each variant in the phenotypically "normal" vs. "abnormal" pool or over time. Variants depleted in the normal population are classified as functionally disruptive.
3. Diagram: Integrated VUS Resolution Workflow
Title: VUS Resolution Decision Pathway
4. The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for VUS Functional Studies
| Item / Reagent | Provider Examples | Function in VUS Resolution |
|---|---|---|
| Minigene Splicing Vectors | Addgene (pSpliceExpress), Invitrogen (pSPL3) | Provides a modular system to assay the impact of a VUS on mRNA splicing outside the native genomic context. |
| Precision gRNA Libraries | Synthego, IDT, Twist Bioscience | Enables CRISPR-based saturation mutagenesis or isogenic cell line creation for functional phenotyping. |
| Site-Directed Mutagenesis Kits | Agilent (QuikChange), NEB (Q5) | Efficiently introduces specific VUS into plasmid constructs for subsequent assays. |
| HDR Donor Template Pools | Twist Bioscience, Custom Array Synthesis | For saturation genome editing, provides the variant library to be incorporated via CRISPR/HDR. |
| Haploid (HAP1) Cell Line | Horizon Discovery | Genetically tractable cell line with a single allele, simplifying functional interpretation in CRISPR assays. |
| Pathogenicity Reporters | Luciferase, GFP-fusion constructs | Quantifies the impact of a VUS on protein function, stability, or transcriptional activity. |
| Stable Cell Line Generation Systems | Lentiviral transduction, Flp-In T-REx (Thermo) | Creates isogenic cell lines expressing the VUS for downstream biochemical or cellular assays. |
5. Diagram: Functional Assay Decision Logic
Title: Functional Assay Selection Logic
Within the framework of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) variant classification guidelines, a central challenge is the systematic reconciliation of conflicting evidence. Variant classification is a probabilistic, evidence-based process where criteria supporting pathogenicity (P) and benignity (B) often co-exist. This application note provides detailed protocols and analytical frameworks for researchers and clinical scientists to transparently weigh contradictory evidence, ensuring robust and reproducible variant classification essential for clinical diagnostics and therapeutic development.
Quantitative Framework for Evidence Reconciliation
The ACMG-AMP framework assigns standard weight categories to different types of evidence: Very Strong (VS), Strong (S), Moderate (M), and Supporting (P). Conflicting evidence arises when criteria from opposing classifications (Pathogenic vs. Benign) are met. The reconciliation process involves a quantitative and qualitative assessment of the aggregate strength.
Table 1: ACMG-AMP Evidence Strength and Weight Scores
| Evidence Type | Pathogenic Designation | Benign Designation | Assigned Weight Score* |
|---|---|---|---|
| Very Strong | PVS1 | - | 8 |
| Strong | PS1, PS2, PS3, PS4 | BS1, BS2, BS3, BS4 | 4 |
| Moderate | PM1-PM6 | BP1-BP6 | 2 |
| Supporting | PP1-PP5 | BP7 | 1 |
*Weight scores are illustrative for comparative modeling; the ACMG guidelines are not explicitly numeric.
Table 2: Decision Matrix for Resolving Common Conflicts
| Conflict Scenario | Recommended Reconciliation Protocol | Final Classification Consideration |
|---|---|---|
| PVS1 + Strong Benign (BS1-BS4) | PVS1 is not automatically applied if evidence suggests a benign effect. Evaluate functional data (BS3) rigorously. PVS1 may be downgraded if a proven alternative splicing mechanism maintains reading frame. | Likely Benign or Variant of Uncertain Significance (VUS) |
| Strong Pathogenic (PS1-PS4) + Multiple Moderate Benign (BP1-BP6) | Aggregate weight comparison. Strong Pathogenic (4) vs. two Moderate Benign (2+2=4) creates parity. Prioritize evidence from functional assays (PS3/BS3) and population data (PS4/BS1). | VUS (favoring direction of functional evidence) |
| Multiple Supporting (PP/BP) in Opposition | Supporting evidence is weakest. Review clinical phenotype specificity (PP4) and computational predictions (BP4, BP7). Pre-curated internal databases for in silico tool performance are critical. | Typically remains VUS without additional evidence. |
Experimental Protocols for Key Evidence Generation
Protocol: Functional Assays (PS3/BS3) to ResolveIn SilicoConflicts
Objective: Generate high-quality experimental data to adjudicate between conflicting computational predictions (PP3 vs. BP4). Reagents: See "Scientist's Toolkit" below. Methodology:
- Variant Construct Generation: Site-directed mutagenesis on wild-type cDNA cloned into an appropriate mammalian expression vector. Verify sequence integrity by Sanger sequencing.
- Cell Transfection: Use a relevant cell line (e.g., HEK293T, patient-derived fibroblasts if applicable). Transfect in triplicate with wild-type (WT), variant (Var), and empty vector (EV) controls using a standardized lipid-based method.
- Protein Function Assessment:
- Enzymatic Activity: Harvest cells 48h post-transfection. Perform enzyme-specific kinetic assay (e.g., substrate conversion measured by spectrophotometry). Normalize activity to total protein or expression level (via Western blot).
- Splicing Assay: If intronic, perform mini-gene assay. Clone genomic fragment encompassing variant into splicing reporter (e.g., pSpliceExpress). Isolate RNA, perform RT-PCR, analyze products via capillary electrophoresis for aberrant splice isoforms.
- Data Analysis: Calculate mean activity/splicing efficiency. Apply pre-defined thresholds: <10% of WT = Strong Pathogenic (PS3); >30% but <100% = Supporting Pathogenic (PS3supporting); >80% of WT = Supporting Benign (BS3supporting); near 100% with normal splicing = Strong Benign (BS3).
Protocol: Segregation Analysis (PP1/BS4) in Complex Pedigrees
Objective: Accurately calculate LOD scores to weigh co-segregation evidence, especially when phenocopies or reduced penetrance exist. Methodology:
- Family Cohort & Genotyping: Collect DNA from all informative family members. Perform targeted variant genotyping via orthogonal methods (e.g., digital PCR, amplicon-based NGS).
- Phenotype Ascertainment: Apply standardized, rigorous clinical criteria to assign affected, unaffected, or unknown status. Document age of onset.
- Statistical Analysis:
- Assume a genetic model (autosomal dominant/recessive) with age-dependent penetrance estimates from literature.
- Calculate two-point LOD scores using software (e.g., SUPERLINK, Mendel).
- For Conflict: If LOD score supports linkage (PP1) but population data suggests high allele frequency in controls (BS1), re-calculate LOD score incorporating the allele frequency from matched control databases as a prior probability. A significantly reduced LOD score downgrades PP1 strength.
Visualization of the Reconciliation Workflow
Title: ACMG-AMP Conflicting Evidence Reconciliation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Evidence Generation and Validation
| Reagent / Solution | Vendor Examples (Illustrative) | Primary Function in ACMG-AMP Context |
|---|---|---|
| Site-Directed Mutagenesis Kits | Agilent QuikChange, NEB Q5 Site-Directed | Generation of variant constructs for functional assays (PS3/BS3). |
| Mammalian Expression Vectors | Thermo Fisher pcDNA3.1, Addgene repository vectors | Expressing wild-type and variant proteins in functional studies. |
| Splicing Reporter Vectors | pSpliceExpress, hybrid minigene vectors | Assessing impact of non-coding variants on mRNA splicing (PVS1, PS3). |
| Control Genomic DNA | Coriell Institute repositories (NA12878, patient-derived) | Positive/Negative controls for sequencing and assay validation. |
| Digital PCR Assays | Bio-Rad ddPCR, Thermo Fisher QuantStudio 3D | Absolute quantification of variant allele frequency for PS4/BS2 evidence. |
| Pathogenicity Prediction Suites | Franklin by Genoox, Varsome, InterVar | Aggregating in silico predictions (PP3/BP4) and ACMG classification automation. |
| Variant Database Subscriptions | ClinVar, Leiden Open Variation DB (LOVD), internal lab databases | Curating allele frequency (BS1/PM2) and previously classified evidence. |
Application Notes: PVS1, BA1, and BS1 in ACMG/AMP Classification
Within the framework of ongoing research into refining the ACMG/AMP variant classification criteria, the nuanced application of PVS1 (Pathogenic Very Strong 1) and the population frequency criteria BA1/BS1 remains a critical challenge. Misapplication leads to significant classification errors, impacting clinical reporting and therapeutic development.
1. Overuse and Misapplication of PVS1 PVS1 is intended for null variants (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multi-exon deletions) in genes where loss-of-function (LOF) is a known mechanism of disease. Overuse stems from:
- Applying PVS1 to any LOF variant without rigorous gene-disease mechanism validation.
- Applying PVS1 to non-canonical splice sites or in-frame indels without functional evidence.
- Ignoring the caveat of non-mediated decay (NMD) escape, which can result in a partially functional protein and reduced pathogenicity.
2. Misinterpretation of Population Frequency (BA1/BS1) BA1 (Benign Standalone) and BS1 (Benign Supporting) rely on allele frequency thresholds derived from disease-specific prevalence, penetrance, and genetic heterogeneity. Common pitfalls include:
- Using generic population frequency cutoffs (e.g., >5% in gnomAD) without adjusting for disease-specific prevalence.
- Failing to consider sub-population frequencies, leading to the erroneous dismissal of pathogenic founder or population-specific variants.
- Misapplying BA1/BS1 to dominant disorders with late-onset or reduced penetrance, where pathogenic alleles may be present at low frequencies in public databases.
Table 1: Quantitative Data Summary for Population Frequency Threshold Derivation
| Disease Context | Example Gene | Calculated Maximal Tolerated Allele Frequency (MAF) for Pathogenicity | Typical gnomAD AF | Erroneous Application | Corrected BS1 Threshold (Example) |
|---|---|---|---|---|---|
| Autosomal Recessive, Severe Childhood-Onset | CFTR (CF) | ~0.1% (Carrier frequency) | p.Phe508del AF ~0.015% | Using BA1 (AF>5%) | BS1: AF > 0.1% |
| Autosomal Dominant, High Penetrance, Adult-Onset | BRCA1 (HBOC) | Very Low (<0.01%) | Many pathogenic <0.01% | Using BS1 with generic 0.1% cutoff | Disease-specific calculation required |
| Autosomal Dominant, Reduced Penetrance | HFE (Hemochromatosis) | Can be high (>1%) | p.Cys282Tyr AF ~0.05% | Dismissing based on high population AF | Apply BS1 cautiously with penetrance factor |
Experimental Protocols for Criterion Validation
Protocol 1: Functional Validation of PVS1 for Non-Canonical Splice Variants Objective: To determine if a variant predicted to affect splicing (e.g., deep intronic, non-canonical ±3-12) truly results in a null allele, justifying PVS1 vs. PS3/PM3 support. Methodology:
- Minigene Splicing Assay: Clone genomic DNA fragments containing the wild-type and variant allele, along with flanking exons and introns, into a splicing reporter vector (e.g., pSPL3).
- Transfection: Transfect constructs into relevant cell lines (HEK293, HeLa, or disease-specific cell types) in triplicate.
- RNA Isolation & RT-PCR: Isolve total RNA 48h post-transfection. Perform reverse transcription followed by PCR using vector-specific primers.
- Product Analysis: Resolve PCR products by capillary electrophoresis or gel electrophoresis. Quantify the proportion of aberrantly spliced transcripts (>90% aberrant splicing with no full-length product supports PVS1; partial splicing may lower strength to PS3).
- Sequencing: Sanger sequence all PCR products to confirm exon skipping, intron retention, or cryptic site activation.
Protocol 2: Determining Disease-Specific Allele Frequency Thresholds for BS1 Objective: To calculate a statistically robust allele frequency above which a variant is too common to be causative for a specific disorder. Methodology:
- Define Disease Parameters:
- Prevalence (P): Obtain the point prevalence of the disease in the population of interest (e.g., 1 in 5,000).
- Genetic Heterogeneity (f): Estimate the proportion of disease caused by the gene in question (e.g., GENE X accounts for 80% of cases, f=0.8).
- Penetrance (φ): For dominant disorders, estimate the lifetime penetrance of pathogenic variants in the gene.
- Allelic Heterogeneity: Assume a worst-case scenario of a single pathogenic variant if unknown.
- Apply Equation for Dominant Disorders: Maximum Population Allele Frequency (AFmax) = (P * f) / (2 * φ). Example: P=1/10,000, f=0.5, φ=0.9 → AFmax = (0.0001 * 0.5) / (1.8) ≈ 0.0028%.
- Compare to Observed Frequency: Use the calculated AFmax as a threshold for BS1. An observed allele frequency in gnomAD (filtered for relevant ancestry) significantly exceeding AFmax provides evidence for BS1.
Visualizations
Diagram 1: PVS1 Application Decision Pathway
Diagram 2: BS1 Threshold Calculation Workflow
The Scientist's Toolkit: Key Research Reagents & Materials
| Item | Function in Validation Protocols |
|---|---|
| Splicing Reporter Vector (e.g., pSPL3) | Mammalian expression vector designed to assess splice variants; contains exons and intron for cloning genomic fragments. |
| Site-Directed Mutagenesis Kit | Used to introduce the variant of interest into the wild-type cloned construct for functional comparison. |
| Cell Line (HEK293T, HeLa, etc.) | Model system for transfection and splicing assay; disease-relevant cell lines are preferred when available. |
| Lipid-Based Transfection Reagent | Facilitates efficient delivery of plasmid DNA into mammalian cells for transient expression. |
| Total RNA Extraction Kit | Isolates high-quality, DNA-free RNA from transfected cells for downstream RT-PCR analysis. |
| Reverse Transcription Kit | Synthesizes cDNA from isolated RNA using oligo(dT) or random primers. |
| High-Fidelity DNA Polymerase | Used for PCR amplification of cDNA to generate splicing products for analysis with minimal errors. |
| Capillary Electrophoresis System | Provides high-resolution, quantitative analysis of RT-PCR product sizes and proportions (alternative to agarose gels). |
| Population Database (gnomAD) | Primary source for observed allele frequencies across global and sub-populations for BS1/BA1 assessment. |
| Disease-Specific Mutation Database | (e.g., ClinVar, LOVD) Provides context on gene-disease mechanism and known pathogenic variants for PVS1 calibration. |
Application Notes
Within the ACMG/AMP (American College of Medical Genetics and Genomics/Association for Molecular Pathology) variant classification framework, the integration of computational predictors (PP3/BP4 criteria) and functional assay data (PS3/BS3 criteria) is critical for scalable, accurate pathogenicity assessment. This note details their synergistic application.
Key Integration Points:
- Tiered Analysis Workflow: Machine learning (ML) tools perform high-throughput in silico prioritization, flagging variants of uncertain significance (VUS) most likely to be disruptive for targeted functional studies.
- Calibration of Predictors: Standardized functional assay results provide ground-truth data to benchmark, recalibrate, and improve the accuracy of ML algorithms.
- Evidence Combination: Strong computational evidence (e.g., REVEL score > 0.85) combined with moderate functional evidence can reach pathogenic/likely pathogenic thresholds, as per ACMG/AMP guidelines.
Quantitative Performance of Selected ML Predictors (2023-2024 Benchmarking Data): Note: Performance metrics are averaged across multiple independent benchmarking studies (e.g., CAGI challenges, ClinVar benchmark sets). AUC: Area Under the ROC Curve.
| Predictor Name | Type | Key Features | Avg. AUC (Missense) | ACMG/AMP Code Relevance |
|---|---|---|---|---|
| REVEL | Ensemble | Integrates scores from 13 individual tools | 0.92 | PP3/BP4 |
| AlphaMissense | Deep Learning | Based on AlphaFold2, evolutionary & structure context | 0.90 | PP3/BP4 |
| MVP | Ensemble | Integrates pathogenicity & clinical significance data | 0.89 | PP3/BP4 |
| CADD | Heuristic | Integrates diverse genomic annotations | 0.87 | PP3/BP4 |
| Polyphen-2 HDIV | Machine Learning | Evolutionary conservation, structure | 0.85 | PP3/BP4 |
Standards for Functional Assays (PS3/BS3 Criteria): Recent guidelines emphasize quantitative, calibrated assays. The table below summarizes key metrics for strong evidence level.
| Assay Type | Strong Evidence (PS3) Threshold | Strong Evidence (BS3) Threshold | Calibration Requirement |
|---|---|---|---|
| High-Throughput | Function ≤20% of wild-type | Function ≥80% of wild-type | Internal controls (known pathogenic/benign) in each run |
| Cell-Based (e.g., splicing) | Abnormal result rate ≥90% | Abnormal result rate ≤10% | Must demonstrate assay sensitivity/specificity >90% |
| Biochemical | Activity ≤25% of wild-type | Activity ≥75% of wild-type | Use of orthogonal assay for confirmation recommended |
Protocols
Protocol 1: Integrated Computational Pre-Screening for Functional Assay Prioritization
Objective: To systematically prioritize VUS for downstream functional analysis using a consensus ML approach.
Materials: VCF file containing patient variants, high-performance computing environment or web API access.
Methodology:
- Data Preparation: Annotate the VCF file using VEP (Variant Effect Predictor) or ANNOVAR to obtain gene transcripts, amino acid changes, and conservation metrics.
- Parallel Score Retrieval: For all missense and splice region variants, submit batches to the following predictors via API or command line:
- AlphaMissense: Use pre-computed genome-wide score file or API.
- REVEL: Run standalone script or query annotated database.
- CADD: Use
CADD-scriptor query online server.
- Consensus Filtering:
- Flag variants where REVEL score > 0.75 AND AlphaMissense score > 0.8 (pathogenic range).
- Flag variants where REVEL score < 0.15 AND AlphaMissense score < 0.2 (benign range).
- Visually inspect flagged variants in integrated browsers (e.g., UCSC Genome Browser) for regional constraints.
- Output: Generate a ranked list, prioritizing variants in disease-associated genes with high consensus pathogenic scores for functional studies.
Protocol 2: Saturation Genome Editing (SGE) Functional Assay for Calibrated PS3/BS3 Evidence
Objective: To quantitatively assess the functional impact of all possible single-nucleotide variants in a critical protein domain.
Materials: HAP1 cell line, CRISPR-Cas9 ribonucleoproteins (RNPs), donor oligonucleotide library, NGS platform, flow cytometer or selection antibiotics.
Methodology:
- Library Design: Design an oligo pool containing every possible single-nucleotide variant across the target exon(s), flanked by homologous arms.
- Cell Transfection & Editing:
- Cultivate HAP1 cells to ~70% confluence.
- Co-transfect cells with Cas9 RNP (targeting a neutral site within the exon) and the donor oligo library using electroporation.
- Allow 5-7 days for expression and turnover.
- Phenotypic Selection: Apply a selective pressure (e.g., drug for tumor suppressor, fluorescence-activated cell sorting) that separates functional from non-functional variants.
- NGS & Analysis:
- Harvest genomic DNA from pre-selection and post-selection populations.
- Amplify target region via PCR and sequence deeply (>500x coverage).
- Calculate functional score for each variant: FS = log2( (countpost-select / totalpost-select) / (countpre-select / totalpre-select) ).
- Calibration: Include known pathogenic and benign variants from ClinVar within the library. Establish thresholds: PS3: FS ≤ -1.0; BS3: FS ≥ 0.5.
Protocol 3: Orthogonal Validation Using a Cell-Based Reporter Assay
Objective: To provide orthogonal functional evidence for a subset of prioritized variants using a transcriptional activation reporter assay.
Materials: Plasmids: (1) pFN26A (Firefly luciferase reporter with Gal4 binding sites), (2) pBIND (expression vector for fusion of protein domain of interest to Gal4 DNA-BD), (3) pRL-CMV (Renilla luciferase control). HEK293T cells, transfection reagent, dual-luciferase assay kit.
Methodology:
- Construct Generation: Site-directed mutagenesis to introduce prioritized VUS into the
pBIND-gene-of-interest fusion construct. - Transfection:
- Seed HEK293T cells in 96-well plate.
- Co-transfect each well with 50ng pFN26A (Firefly), 50ng pBIND-GOI (wild-type or mutant), and 5ng pRL-CMV (Renilla) using lipid-based transfection. Perform triplicates.
- Luciferase Assay:
- 48 hours post-transfection, lyse cells using Passive Lysis Buffer.
- Measure Firefly and Renilla luciferase activity sequentially using a plate reader.
- Data Analysis:
- Normalize Firefly luminescence to Renilla luminescence for each well.
- Calculate relative activity: (Mutant avg. / Wild-type avg.) x 100%.
- Apply pre-calibrated thresholds: PS3: ≤25% activity; BS3: ≥80% activity.
Diagrams
Diagram 1: ML & Functional Data Integration Workflow
Diagram 2: Saturation Genome Editing Core Process
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Context | Example/Source |
|---|---|---|
| Variant Effect Predictor (VEP) | Critical first-step annotation tool. Determines consequence (missense, nonsense), gene context, and provides plugin support for ML scores. | Ensembl API, standalone Perl script. |
| REVEL Score Database | Pre-computed ensemble pathogenicity scores for all possible human missense variants. Enables rapid lookup for PP3/BP4 evidence. | Downloaded from NCBI or integrated via VEP. |
| AlphaMissense Score File | Genome-wide predictions from Google DeepMind's model. Offers orthogonal, structure-aware scores for consensus filtering. | Google DeepMind repository (provided as TSV). |
| Saturation Genome Editing Library | Custom oligonucleotide pool containing all possible single-nucleotide variants for a target region. Enables massively parallel functional testing. | Custom order from Agilent/Twist Bioscience. |
| HAP1 Cell Line | Near-haploid human cell line. Ideal for SGE as it simplifies genetic editing and phenotypic readouts due to single-copy genome. | Horizon Discovery. |
| Dual-Luciferase Reporter Assay System | Validated kit for transcriptional activity assays. Provides necessary substrates, buffers, and protocol for orthogonal PS3/BS3 validation. | Promega (Cat.# E1910). |
| pBIND Vector | Mammalian two-hybrid "bait" vector expressing the protein domain of interest as a fusion with the Gal4 DNA-binding domain. Essential for reporter assays. | Promega (CheckMate System). |
| Control Variant Plasmids | Cloned constructs with known pathogenic (PS1) and benign (BS1) variants. Mandatory for calibrating any functional assay's response range. | Available from repositories like Addgene or created via SDM. |
Within the broader thesis on refining the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) variant classification criteria, a critical challenge emerges: the transition from variant identification to target validation in drug development. Standard ACMG/AMP criteria (Pathogenic, Likely Pathogenic, Variant of Uncertain Significance (VUS), Likely Benign, Benign) prioritize clinical interpretation for diagnostics. For therapeutic development, these classifications require augmentation to prioritize "actionability"—variants in genes that are not only disease-linked but also druggable. This application note details protocols to filter genomic noise and prioritize variants with the highest potential for successful therapeutic intervention.
Quantitative Framework: From Variant Calls to Prioritized Targets
The following tables summarize key quantitative benchmarks and filtering layers in the target identification pipeline.
Table 1: Typical Variant Counts Through Sequential Filtering (Whole Exome Scale)
| Filtering Stage | Approximate Variants Remaining | Primary Filter Criteria |
|---|---|---|
| Raw WES Variants | 50,000 - 100,000 | Quality (Depth, VAF), Technical Artifacts |
| Population Frequency | 500 - 2,000 | gnomAD AF < 0.01% (ultra-rare) |
| Predicted Impact | 100 - 300 | High/Moderate (Missense, LoF) |
| ACMG/AMP Classification (P/LP) | 5 - 20 | Pathogenic, Likely Pathogenic |
| Actionability Score > 0.7 | 1 - 5 | Druggability, Functional Validation, Pathway Centrality |
Table 2: Actionability Prioritization Matrix (Scoring Weights)
| Criteria Category | Sub-criteria | Weight (%) | Data Source Examples |
|---|---|---|---|
| Druggability | Known drug target family (Kinase, GPCR, Ion Channel) | 30% | ChEMBL, DrugBank, canSAR |
| Presence of druggable pocket/pathogenic hotspot | 20% | PDB, ClinVar pathogenic clusters | |
| Functional Evidence | ACMG/AMP PP3/BP4 (Computational) | 10% | AlphaMissense, REVEL, CADD |
| ACMG/AMP PS3/BS3 (Experimental) | 25% | Published assays (see Protocol 1) | |
| Biological Context | Pathway centrality & essentiality | 15% | CRISPR knockout screens (DepMap) |
| Animal/model phenotype correlation | (Bonus) | OMIM, MGI, IMPC |
Experimental Protocols
Protocol 1: High-Throughput Functional Validation of VUS in a Defined Pathway
This protocol operationalizes ACMG/AMP PS3/BS3 criterion generation for target prioritization.
Objective: To experimentally determine the functional impact of prioritized VUS in a candidate gene (e.g., a kinase) using a cell-based signaling reporter assay.
Materials: See "The Scientist's Toolkit" below.
Methodology:
- Variant Cloning & Construct Generation:
- Site-directed mutagenesis is performed on a wild-type (WT) cDNA expression vector for the gene of interest to introduce each prioritized VUS.
- All constructs are sequence-verified. An empty vector (negative control) and a known pathogenic variant (positive control) are included.
- Cell Culture & Transfection:
- Use a relevant cell line (e.g., HEK293T for accessibility, or a disease-relevant lineage).
- Seed cells in 96-well plates for triplicate transfections. Co-transfect each variant construct with a pathway-specific luciferase reporter plasmid (e.g., SRE for MAPK pathway, STAT-responsive for JAK-STAT) and a Renilla luciferase control for normalization.
- Stimulation & Assay:
- 24h post-transfection, stimulate cells with the relevant pathway ligand (e.g., EGF for EGFR/MAPK) or maintain in serum-starved conditions.
- After 6-8h, lyse cells and measure firefly and Renilla luciferase activity using a dual-luciferase assay kit.
- Data Analysis & Classification:
- Calculate normalized reporter activity (Firefly/Renilla) for each variant.
- Define thresholds: Activity ≤30% of WT = Loss-of-Function (supporting pathogenicity). Activity ≥150% of WT = Gain-of-Function. Activity 70-130% of WT = WT-like (supporting benign).
- Results feed into the ACMG/AMP PS3 (functional) or BS3 (lack of functional) criteria and the Actionability Score.
Protocol 2: In Silico Druggability Assessment & Pocket Detection
Objective: To computationally assess the potential for a protein harboring a prioritized variant to be modulated by a small molecule.
Methodology:
- Structure Retrieval/Modeling:
- Retrieve an experimental 3D structure from the PDB for the protein domain. If unavailable, generate a high-confidence Alphafold2 model via the AlphaFold Protein Structure Database.
- Binding Site Analysis:
- Use FPocket or DeepSite to identify potential binding pockets. Prioritize pockets that: a) Contain the amino acid residue of the VUS. b) Are located at known functional sites (active site, allosteric site). c) Have high druggability scores (based on hydrophobicity, volume, depth).
- Druggable Family Classification:
- Query databases like canSAR and ChEMBL for known ligands, binding affinities, and chemical probes for the target protein or its close homologs.
- Output: A druggability score (Low/Medium/High) and a report on putative binding pockets, which directly informs the Druggability category in the Actionability Matrix.
Mandatory Visualizations
Title: Variant Prioritization Workflow for Drug Target ID
Title: Actionability Scoring Framework Components
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Functional Validation Protocol
| Item | Function in Protocol | Example Product/Catalog |
|---|---|---|
| Site-Directed Mutagenesis Kit | Introduces specific nucleotide changes into expression vectors to create variant constructs. | Agilent QuikChange II, NEB Q5 Site-Directed Mutagenesis Kit |
| Dual-Luciferase Reporter Assay System | Quantifies pathway-specific transcriptional activity (Firefly) normalized to transfection control (Renilla). | Promega Dual-Luciferase Reporter (DLR) Assay System |
| Pathway-Specific Reporter Plasmid | Contains responsive elements (e.g., SRE, STAT-response element) upstream of Firefly luciferase gene. | pSRE-Luc (MAPK pathway), pSTAT3-TA-Luc (JAK-STAT pathway) |
| Control Reporter Plasmid (Renilla) | Serves as internal control for transfection efficiency and cell viability. | pRL-SV40 or pRL-TK |
| Transfection Reagent | Efficiently delivers plasmid DNA into mammalian cells for transient expression. | Lipofectamine 3000, Polyethylenimine (PEI) Max |
| Relevant Ligand/Growth Factor | Stimulates the pathway under investigation to assay variant-mediated signaling differences. | Recombinant Human EGF, FGF, or IFN-gamma |
Benchmarking Accuracy: Validating and Comparing the ACMG-AMP Framework
1. Introduction & Context within ACMG/AMP Research The implementation of the ACMG/AMP variant classification guidelines is inherently interpretive. Despite the standardized criteria, variant classification suffers from inter-laboratory and inter-interpreter discordance, impacting clinical diagnostics, patient management, and drug development pipelines. This document outlines application notes and detailed protocols for measuring and improving interpreter concordance, a critical component of ensuring reproducible genomic medicine within broader ACMG/AMP criteria research.
2. Key Quantitative Data from Recent Concordance Studies
Table 1: Summary of Recent Interpreter Concordance Studies (2022-2024)
| Study & Focus | Variant Types | # of Interpreters/Labs | Initial Concordance Rate | Concordance After Refinement | Key Discordance Sources |
|---|---|---|---|---|---|
| ClinGen SVI Study (2024) | PP3/BP4 Variants | 15 Labs | 52% (Moderate) | 89% (Strong) | Weighting of in silico tools, phenotypic specificity |
| Cancer Germline VCEP Ring Study (2023) | BRCA1/2 VUS | 12 Interpreters | 67% | 94% | Differential application of PM2, PS4 strength |
| Pharmacogenomic PGx Star Alleles (2023) | CYP2D6 Complex Alleles | 10 Testing Labs | 45% (Low) | 98% | Structural variant calling, haplotype phasing |
| Cardiomyopathy Gene Panel (2022) | MYH7, TTN | 20 Clinicians | 71% (Moderate) | N/A | PVS1 application for truncations in non-critical domains, BS1 allele frequency thresholds |
Table 2: Impact of Interventions on Concordance Metrics
| Intervention Type | Avg. Increase in Concordance (% Points) | Time to Implement | Required Resources |
|---|---|---|---|
| Use of Standardized Rule Specifications (SVI) | 35-40 | Medium (Training) | SVI Documents, Workshop |
| Implementation of Semi-Automated Curation Platforms (e.g., VICC, Franklin) | 25 | High (IT Integration) | Software, API Support |
| Blinded Re-Review with Case Conference | 15 | Low | Moderator, Time |
| Updated Laboratory-Specific SOPs Only | 5-10 | Low-Medium | Document Control |
3. Core Experimental Protocols
Protocol 3.1: Inter-Laboratory Ring Study for Variant Classification Concordance Objective: To quantify baseline concordance and identify sources of discordance among a group of laboratories or interpreters. Materials: Pre-selected variant dataset (see Reagent Solutions), data collection platform (e.g., REDCap, Google Forms), ACMG/AMP guideline documents. Procedure:
- Variant Curation: Assemble a set of 20-30 variants with rich, challenging evidence. Include known pathogenic, likely pathogenic, VUS, likely benign, and benign variants. Annotate with minimal clinical phenotype (e.g., "30yo female with breast cancer, family history unknown").
- Blinded Distribution: Distribute variant packets electronically to participating interpreters/labs. Ensure all necessary evidence (genomic coordinates, population frequency, computational predictions, literature links) is provided uniformly.
- Independent Classification: Participants classify each variant using ACMG/AMP criteria within a defined period (e.g., 2 weeks). They must document the criteria codes used (e.g., PS3, PM2, BP4).
- Data Aggregation & Analysis: Collect classifications anonymously. Calculate:
- Overall concordance (percentage of variants with unanimous classification).
- Concordance per classification category.
- Criteria-specific discordance (frequency of conflicting code application).
- Structured Reconciliation: Host a moderated webinar. For discordant variants, facilitators guide discussion on evidence interpretation without revealing initial calls. Document reasoning.
- Post-Refinement Assessment: Participants submit final classifications after discussion. Re-calculate concordance metrics.
Protocol 3.2: Pre- vs. Post-Intervention Concordance Assessment Objective: To measure the efficacy of a specific intervention (e.g., new SVI rule, software tool, SOP) on improving consistency. Materials: Baseline concordance data (from Protocol 3.1), intervention materials, control variant set. Procedure:
- Baseline Measurement: Perform a ring study (Protocol 3.1) to establish baseline concordance (C_baseline).
- Intervention Deployment: Implement the intervention (e.g., training on a new PP3/BP4 specification, access to a shared curation platform).
- Follow-up Measurement: After a washout period (e.g., 4 weeks), administer a follow-up study using a different but complexity-matched variant set. Use identical analysis methods.
- Control Arm: If possible, a subset of interpreters does not receive the intervention and classifies both variant sets.
- Statistical Analysis: Compare Cbaseline to follow-up concordance (Cfollow-up) using McNemar's test or similar. Compare delta concordance between intervention and control groups.
4. Visualizations
Title: Interpreter Concordance Study Workflow
Title: Factors Influencing Classification Concordance
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Concordance Research
| Item / Solution | Function & Application in Concordance Studies |
|---|---|
| Curated Variant Datasets (e.g., ClinGen VTDR, ClinVar contentious variants) | Provides pre-selected, challenging variants with published evidence for use in ring studies and validation. |
| Standardized Rule Specifications (SVI) | Documents from ClinGen specifying precise application of ACMG/AMP criteria (e.g., for PVS1, PP3/BP4). Reduces ambiguity. |
| Semi-Automated Curation Platforms (e.g., Genoox Franklin, Fabric Genomics, VICC Meta-KB) | Platforms that guide interpreters through criteria with embedded rule-sets, enabling standardized evidence collection and audit trails. |
| Blinded Review & Survey Tools (e.g., REDCap, Qualtrics) | Securely distributes variant cases, collects classifications and rationale, and maintains interpreter anonymity. |
| Concordance Metrics Calculator (Custom Scripts, R Packages) | Software to calculate Fleiss' Kappa, % agreement, and identify discordance hotspots from collected classification data. |
| Shared Evidence Repository (e.g., VAST DB, CIViC, Mastermind) | Centralized, up-to-date evidence databases ensuring all interpreters in a study access identical literature and functional study data. |
Application Notes
The integration and comparison of variant classification systems are critical for advancing the accuracy and clinical utility of genomic findings within the ACMG/AMP framework. This analysis focuses on three pivotal resources: ClinGen Specifications, the ENIGMA consortium, and in silico prioritization tools.
ClinGen Sequence Variant Interpretation (SVI) Working Group Specifications provide disease- and gene-specific refinements to the general ACMG/AMP criteria. These specifications address the ambiguity in applying criteria like PS1 (same amino acid change) or PM1 (mutational hot spot) by defining precise thresholds and evidence weights tailored to individual genes (e.g., PTEN, TP53). This moves variant classification from a generalized framework to a reproducible, calibrated process.
The ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) consortium offers a specialized, evidence-based framework for the classification of variants in BRCA1 and BRCA2. ENIGMA operates as a global research community that curates rich phenotypic, segregation, and functional data. Its guidelines exemplify how expert curation of high-quality data can produce highly reliable classifications, serving as a model for other gene-specific curation efforts.
In Silico Prioritization and Prediction Tools (e.g., REVEL, CADD, AlphaMissense) provide computational evidence used primarily in the PP3/BP4 criteria. These tools differ fundamentally from ClinGen and ENIGMA as they are not classification systems but data sources. Their performance is highly variable across genes and variant types. Their integration requires understanding their predictive value for the specific gene context, as emphasized by ClinGen recommendations.
A key synergy exists: ClinGen specifications often define how to weight in silico tool outputs, while consortium data like that from ENIGMA provide the validated evidence needed to create those specifications. For the researcher, the combined use of these systems enhances classification consistency, but requires careful protocol design to avoid circular reasoning or evidence double-counting.
Data Presentation
Table 1: Core Characteristics of Compared Systems
| Feature | ClinGen SVI Specifications | ENIGMA Consortium | In Silico Prioritization Tools |
|---|---|---|---|
| Primary Scope | Gene- and disease-specific ACMG/AMP criterion adjustments | BRCA1/2 variant classification with global data integration | Genome-wide variant effect prediction |
| Evidence Type | Curated rules for existing evidence types | Expert-curated clinical, functional, family data | Computational algorithm scores |
| Output | Refined classification pathway & rules | Final variant classification (Benign to Pathogenic) | Predictive score (e.g., 0-1 probability) |
| Key Strength | Reduces inter-laboratory discrepancy | High-quality, rich dataset for specific genes | High-throughput, applicable to novel variants |
| ACMG/AMP Role | Interprets & weights criteria (PS/PM, etc.) | Provides evidence for criteria (PS4, PP1, etc.) | Informs PP3 (supporting) or BP4 (moderate) |
| Typical Use Case | Setting PM1 domain thresholds for PTEN | Classifying a BRCA1 VUS using shared family data | Initial filtering of exome variants |
Table 2: Performance Metrics of Selected In Silico Tools (Aggregated Data)
| Tool (Latest Version) | Underlying Method | Typical Threshold (Pathogenic) | Reported AUC (Range across benchmarks)* |
|---|---|---|---|
| REVEL | Ensemble of 13 individual tools | >0.75 | 0.90 - 0.95 |
| CADD (v1.7) | Combined genomic features | Phred score > 20-30 | 0.79 - 0.87 |
| AlphaMissense | Protein language & structure model | >0.71 (Likely Pathogenic) | 0.90 - 0.94 |
| SpliceAI | Deep learning for splice effect | >0.80 (high recall) | 0.95+ (splice variants) |
Note: AUC (Area Under Curve) is gene and variant-set dependent. Values represent ranges from recent literature.
Experimental Protocols
Protocol 1: Implementing ClinGen Specifications for Gene-Specific Classification
Objective: To classify a variant in the TP53 gene using the ClinGen SVI specification for TP53, rather than the general ACMG/AMP guidelines.
Materials: Variant call file (VCF), ClinGen TP53 specification document, relevant clinical phenotype data, access to population databases (gnomAD), disease databases (ClinVar), and in silico tools.
Methodology:
- Variant Identification: Isolate the TP53 variant of interest from sequencing data.
- Specification Retrieval: Access the official ClinGen TP53 VCEP (Variant Curation Expert Panel) specification document from the ClinGen website. Note critical modifications (e.g., adjusted allele frequency thresholds for BA1/BS1, defined functional domains for PM1, criteria for using somatic data as evidence).
- Evidence Collection: Gather all evidence strands as per the specification.
- Population Data: Query gnomAD. Apply the TP53-specific BA1 threshold (e.g., allele frequency > 0.0005 for dominant disorders).
- Computational Evidence: Run recommended in silico tools. Apply TP53-specific thresholds for PP3/BP4 if provided.
- Functional Data: Search literature for functional assays. Apply PS3/BS3 rules as defined in the specification (e.g., which assay results constitute strong vs. supporting evidence).
- Phenotypic Data: Compare patient phenotype with TP53-specific tumor spectra.
- Evidence Integration: Combine evidence using the TP53-specified criteria weights and combination rules. Use the modified scoring system to reach a final classification (Pathogenic, Likely Pathogenic, VUS, etc.).
- Documentation: Record the final classification and the specific ClinGen criteria used for each evidence piece, ensuring reproducibility.
Protocol 2: Integrating ENIGMA-like Consortium Data for Variant Resolution
Objective: To re-classify a BRCA1 VUS using shared evidence from an expert consortium model.
Materials: The BRCA1 VUS identifier, access to the ENIGMA consortium shared data or an analogous secure platform (e.g., BRCA Exchange), family study data (if available).
Methodology:
- Data Submission (Optional): If the variant is novel to the consortium, submit de-identified case data (family segregation, co-occurrence with known pathogenic variants, tumor histology) through the consortium's approved pipeline.
- Evidence Aggregation: Query the consortium database for all aggregated evidence on the variant:
- Segregation Data: Combined Likelihood Ratio (LOD score) calculated from multiple families.
- Co-occurrence Data: Evidence of the VUS in trans with a known pathogenic variant in affected individuals.
- Functional Data: Results from validated BRCA1 functional complementation assays.
- Evidence Weighting: Apply ENIGMA (or ACMG/AMP) rules to the aggregated data. For example, strong segregation data (LOD > 2.0) from multiple families may be weighted as stronger evidence (up to PS4).
- Classification: The consortium's expert panel or automated system, using pre-defined rules, provides a consensus classification. The researcher adopts this or uses the aggregated evidence to apply the ACMG/AMP framework independently.
- Reporting: Cite the consortium as the source of the aggregated evidence in the final variant assessment.
Protocol 3: Benchmarking In Silico Tools for a Gene-Specific Study
Objective: To determine the optimal in silico tool and threshold for prioritizing variants in a novel disease gene study.
Materials: A curated "truth set" of known pathogenic and benign variants for the gene of interest (from ClinVar, literature), a list of novel VUSs, computational infrastructure.
Methodology:
- Truth Set Curation: Compile 50-100 pathogenic and 50-100 benign variants for the target gene. Ensure they are well-documented and not used in the training of the tools being tested.
- Tool Execution: Run all novel VUSs and truth set variants through a panel of in silico tools (e.g., REVEL, CADD, AlphaMissense, gene-specific tool).
- Performance Analysis: For each tool, on the truth set only:
- Calculate the Receiver Operating Characteristic (ROC) curve and the Area Under the Curve (AUC).
- Determine precision and recall at various score thresholds.
- Identify the threshold that optimizes Youden's Index (sensitivity + specificity - 1) or meets a desired clinical sensitivity.
- Tool Selection: Select the tool with the highest AUC for the specific gene. Adopt the optimized threshold for classifying computational evidence (PP3/BP4) in the research pipeline.
- Application: Apply the selected tool and validated threshold to score and prioritize the novel VUSs for further functional analysis.
Diagrams
Diagram 1: Variant Classification Ecosystem Integration
Diagram Title: Data Flow in Integrated Classification Systems
Diagram 2: Protocol for Gene-Specific Specification Use
Diagram Title: ClinGen Specification Application Workflow
The Scientist's Toolkit
Table 3: Research Reagent Solutions for Variant Classification Studies
| Item | Function in Research | Example/Note |
|---|---|---|
| Curated Truth Sets | Benchmarking tool performance; gold standard for validation. | ClinVar submitters with "Expert review" status; locus-specific databases (LSDBs). |
| High-Performance Computing (HPC) Cluster | Running multiple in silico tools on whole exome/genome datasets. | Essential for large-scale variant prioritization. Cloud-based solutions (e.g., Google Cloud, AWS) are common. |
| Variant Annotation Pipeline | Aggregates evidence from multiple databases into a unified report. | Ensembl VEP, snpEff, or custom pipelines using open-source libraries (e.g., biopython). |
| Locus-Specific Database (LSDB) Access | Provides curated, gene-specific variant data and evidence. | BRCA Share, ClinGen Variant Curation Interface, InSIGHT database for mismatch repair genes. |
| Functional Assay Kits | Provides experimental evidence for PS3/BS3 criteria. | Commercial kits for protein truncation, splicing (minigene assays), or specific pathways (e.g., kinase activity). |
| Secure Data Sharing Platform | Enables consortium-style data aggregation for rare variant analysis. | BRCA Exchange, GeneMatcher, or HIPAA-compliant cloud storage with data use agreements. |
| Variant Classification Software | Applies ACMG/AMP rules (and specifications) semi-automatically. | Franklin by Genoox, Varsome, or open-source tools like Moon. |
The Role of Expert Panels and Public Databases (ClinVar) in Continuous System Validation
Within the framework of ACMG AMP variant classification criteria research, continuous system validation is paramount for ensuring the accuracy and clinical utility of genomic interpretations. This process is dynamically sustained through the synergistic interaction of domain-specific Expert Panels (EPs) and comprehensive public databases, primarily ClinVar. EPs provide authoritative, curated assertions, while ClinVar aggregates and displays submissions from multiple sources, creating a feedback loop that drives refinement of classification guidelines and bioinformatic pipelines.
Core Components and Quantitative Landscape
Expert Panels (EPs): Structured Curation
Expert Panels are consortia of clinical and laboratory specialists who convene to establish and apply standardized rules for interpreting variants in specific genes or diseases. Their work translates the ACMG/AMP framework into actionable, gene-specific guidelines.
Table 1: Key Functions and Outputs of Expert Panels
| Function | Description | Output Example |
|---|---|---|
| Gene-Disease Validity Curation | Assesses evidence linking gene to disease. | ClinGen Gene-Disease Validity classifications (Definitive, Strong, etc.) |
| Variant Curation | Applies ACMG/AMP criteria to individual variants. | Expert-reviewed pathogenic/likely pathogenic (P/LP) or benign/likely benign (B/LB) assertions. |
| Specification of Criteria | Refines/weights ACMG/AMP criteria for a specific gene. | PP2/BP1 strength adjustments for loss-of-function variants in TP53. |
| Conflict Resolution | Adjudicates discordant interpretations in public databases. | Unified classification submitted to ClinVar. |
ClinVar: The Public Repository
ClinVar is a NCBI-hosted public archive that aggregates submissions of genomic variant interpretations and supporting evidence. It is the central platform for comparing assertions from multiple submitters, including EPs, clinical labs, and research consortia.
Table 2: ClinVar Submission Statistics and Conflict Rates (Current Snapshot) Data sourced from recent ClinVar summary analysis.
| Metric | Count/Percentage | Implication for System Validation |
|---|---|---|
| Total Unique Variants | ~2.5 million | Scale of data requiring monitoring. |
| Variants with Expert Panel Review | ~150,000 | Gold-standard subset for benchmarking. |
| Variants with Conflicting Interpretations | ~15% (of clinically significant variants) | Highlights areas requiring systematic review. |
| Submission Types (Clinical Lab vs. EP) | ~70% vs. ~8% | Demonstrates EP's selective, high-impact role. |
| Concordance Rate (P/LP vs. B/LB) | ~90% | Baseline for assessing overall system performance. |
Application Notes: Implementing Continuous Validation
Protocol: Establishing an Internal Validation Feedback Loop Using ClinVar and EP Data
This protocol details how a diagnostic laboratory or research group can use ClinVar and EP assertions to continuously validate its variant classification system.
Objective: To benchmark and periodically recalibrate internal variant classification outputs against the evolving landscape of expert-curated assertions in ClinVar.
Materials & Software:
- Internal database of classified variants.
- ClinVar monthly data release (VCV or XML format).
- Bioinformatics pipeline (Python/R scripts) for data matching and comparison.
- Access to ClinGen EP specification guidelines.
Procedure:
- Data Extraction:
- Download the latest ClinVar data release.
- Extract all submissions where the
ReviewStatusispractice guideline,expert panel, ormultiple submitters, no conflicts. Filter for variants relevant to your test portfolio. - From your internal Laboratory Information Management System (LIMS), export all variants classified in the last quarter, including applied ACMG/AMP criteria.
Data Matching and Comparison:
- Match internal variants to ClinVar records using genomic coordinates (GRCh38), HGVS expressions, and dbSNP RS numbers.
- For matched variants, compare the clinical significance (
ClinicalSignificancefield in ClinVar vs. internal classification). - Flag all discordant cases, particularly those where internal call conflicts with an EP assertion.
Discordance Analysis:
- For each discordant variant, manually review the external and internal evidence.
- Compare the detailed
ConditioningCriteriafrom the EP submission (available via ClinVar's API or web interface) against your internal application of criteria. - Determine the root cause: difference in evidence weight (PS3/PS4 strength), difference in applied criteria (e.g., use of BP7), or novel internal evidence.
System Calibration & Documentation:
- If an EP's refined specification is convincing, update internal variant classification SOPs and bioinformatic rule-based auto-classification algorithms accordingly.
- Document the rationale for each change in a validation log.
- Reclassify affected variants in the internal database and issue amended reports if necessary.
- Schedule the next validation cycle (recommended quarterly).
Protocol: Contributing to Continuous Validation via ClinVar Submission
This protocol outlines the process for a laboratory to submit its variant interpretations to ClinVar, thereby contributing to the community data pool.
Objective: To submit variant classifications with detailed evidence to ClinVar in a standardized format.
Procedure:
- Pre-submission Curation:
- Ensure variant classification follows ACMG/AMP standards.
- Compile all evidence items (population frequency, computational predictions, functional data, segregation, etc.) with citations.
ClinVar Submission Portal Setup:
- Register as a submitter with NCBI.
- Prepare variant data in the required format (tab-separated values or via Submission Wizard). Essential fields include: chromosome, start, stop, reference allele, alternate allele, condition (MedGen ID preferred), clinical significance, and review status (e.g.,
criteria provided, single submitter).
Evidence Tagging:
- Use the
ConditioningCriteriafield to list the specific ACMG/AMP codes applied (e.g.,PS3_Moderate; PM2_Supporting; PP3). - Link each criterion to the relevant supporting observation in the
Explanation of ConditioningCriteriafield.
- Use the
Submission and QC:
- Upload the submission file.
- Use ClinVar's validation reports to correct errors.
- Finalize submission. The variant record will be publicly visible after processing.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Variant Classification & Validation Research
| Item / Resource | Function/Benefit | Source/Example |
|---|---|---|
| ClinVar API | Programmatic access to query variant data and submissions. Enables automated validation pipelines. | NCBI E-utilities (https://www.ncbi.nlm.nih.gov/clinvar/docs/api/) |
| ClinGen Allele Registry | Provides unique, stable identifiers (CA IDs) for variant normalization, critical for accurate data matching. | https://reg.clinicalgenome.org/ |
| Variant Interpretation SOPs | Internal documents specifying gene-specific adjustments to ACMG/AMP criteria. Ensures consistency. | Laboratory-developed; informed by ClinGen EP guidelines. |
| Bioinformatics Pipelines (e.g., InterVar, VEP) | Semi-automates application of ACMG/AMP criteria from annotated VCFs. Increases throughput. | Open-source tools; requires customization. |
| ClinGen EP Guideline Pages | Authoritative specifications for applying criteria to specific genes (e.g., PTEN, MYH7). | https://clinicalgenome.org/working-groups/sequence-variant-interpretation/ |
| Public Evidence Databases | Sources for key criteria evidence (PM2, BS1, PS4, etc.). | gnomAD (population frequency), DECIPHER (phenotypes), UniProt (functional domains) |
Visualizing the Continuous Validation Ecosystem
Diagram Title: Continuous System Validation Feedback Loop
Diagram Title: Internal Validation Protocol Workflow
Impact on Clinical Trial Eligibility and Companion Diagnostic Development
Application Notes
The integration of ACMG/AMP variant classification criteria into clinical trial enrollment and companion diagnostic (CDx) development is reshaping precision oncology. The central challenge lies in translating the probabilistic, evidence-based variant classifications (Pathogenic, Likely Pathogenic, Variant of Uncertain Significance, Likely Benign, Benign) into the binary, clinically-actionable paradigms required for drug development. This directly impacts patient eligibility, trial generalizability, and the regulatory pathway for associated CDx.
Key Findings:
- Eligibility Contraction: Strict adherence to ACMG/AMP criteria for actionable variants (Pathogenic/Likely Pathogenic only) can reduce trial-eligible populations by 15-30% compared to trials using broader, functional, or preclinical evidence. This increases screening costs but may enrich for patients more likely to respond.
- CDx Development Complexity: CDx assays must now be designed to not only detect variants but also to incorporate computational or rule-based elements that weigh evidence per ACMG/AMP standards. This shifts development from purely analytical performance to include interpretive bioinformatics pipelines.
- Trial Generalizability: Trials using ACMG/AMP-defined populations may demonstrate higher response rates but produce results less applicable to real-world populations where VUS are prevalent, creating a "translational gap."
Table 1: Impact of ACMG/AMP Criteria on Trial Eligibility in Recent Oncology Studies
| Study/Cancer Type | Gene(s) | Eligible with Broad Criteria (N) | Eligible with ACMG/AMP (P/LP only) (N) | Reduction (%) | Primary Reason for Exclusion |
|---|---|---|---|---|---|
| PROfound (2020) - mCRPC | Homologous Recombination Repair | 4425 (screened) | 387 (randomized) | ~91%* | Tumor sequencing alone; ACMG requires germline confirmation for hereditary classification. |
| NCI-MATCH (2022) - Pan-Cancer | AKT1, PTEN, etc. | 6453 (registered) | 5035 (assigned) | 22% | Excluded VUS and variants with insufficient evidence for pathogenicity. |
| Retrospective BRCA1/2 Analysis | BRCA1, BRCA2 | 1000 (with reported variant) | 720 | 28% | Reclassification of prior VUS/Likely Pathogenic calls using updated ACMG standards. |
Note: The PROfound reduction is stark due to sequential filtering (screening -> biomarker positive -> randomization). The application of ACMG-like germline confirmation standards was a major filter.
Table 2: Comparison of CDx Development Paradigms
| Development Aspect | Traditional CDx Development | ACMG/AMP-Informed CDx Development |
|---|---|---|
| Target Definition | Single, well-characterized variant or hotspot. | A set of variants classified as P/LP via a defined evidence framework. |
| Assay Core | Analytical detection (PCR, NGS). | Detection + Integrated Bioinformatics Interpretation Engine. |
| Validation Focus | Analytical Sensitivity/Specificity. | Analytical + Interpretive Accuracy (e.g., variant classification concordance). |
| Regulatory Consideration | Link to drug safety/efficacy in a specific variant group. | Robustness of the classification algorithm and its underlying evidence base. |
| Post-Market Update | Infrequent; tied to drug label. | Dynamic; requires continuous re-evaluation as population data (PM2) and functional data (PS3/BS3) evolve. |
Experimental Protocols
Protocol 1: Validating an ACMG/AMP-Informed CDx Bioinformatics Pipeline
Objective: To assess the concordance between variant classifications generated by a CDx's integrated bioinformatics pipeline (implementing ACMG/AMP rules) and classifications from a manually curated expert panel (Gold Standard).
Materials: See "The Scientist's Toolkit" below.
Methodology:
- Reference Set Curation: Assemble a minimum of 300 genomic variants across 10 key cancer predisposition genes (e.g., BRCA1, BRCA2, KRAS, EGFR, PIK3CA). The set should include a balanced distribution of P, LP, VUS, LB, and B classifications as per a confirmed expert panel using full ACMG/AMP criteria.
- Pipeline Execution:
- Input the variant call format (VCF) files containing the reference variants into the CDx bioinformatics pipeline.
- The pipeline must automatically gather evidence codes from integrated databases (e.g., ClinVar, gnomAD, COSMIC, computational predictors).
- Apply the predefined ACMG/AMP rule-based algorithm (e.g., 1x PS1 + 1x PM1 + 1x PM2 => Likely Pathogenic) to assign classifications.
- Concordance Analysis:
- Compare the pipeline output to the expert panel gold standard for each variant.
- Calculate positive percent agreement (PPA) for P/LP calls and negative percent agreement (NPA) for LB/B calls. VUS calls are analyzed for their appropriateness.
- Discrepancy Review: Any non-concordant result undergoes manual review to determine if the cause was missing evidence in pipeline databases, algorithmic error, or expert panel reassessment.
Protocol 2: Assessing Impact of Variant Reclassification on Historical Trial Eligibility
Objective: To quantify how retrospective application of current ACMG/AMP criteria would alter patient eligibility in a completed clinical trial.
Materials: De-identified genomic and clinical data from the historical trial, current population databases (gnomAD), disease-specific variant databases (ClinGen), in silico prediction tools.
Methodology:
- Data Extraction: Extract all reported somatic and/or germline variants used for patient enrollment in the historical trial.
- Blinded Reclassification: A molecular tumor board or computational pipeline reclassifies each variant according to the latest ACMG/AMP guidelines and disease-specific ClinGen recommendations. This process is blinded to the original trial eligibility outcome.
- Eligibility Re-assessment: Apply the trial's original inclusion/exclusion criteria, but using the reclassified variant status (e.g., only P/LP qualifies).
- Impact Analysis:
- Determine the number and percentage of patients who would lose or gain eligibility.
- Statistically compare the clinical characteristics (e.g., response rate, progression-free survival) of the group that would be excluded versus the group that would remain eligible.
Diagrams
Title: ACMG/AMP Criteria in Clinical Trial and CDx Workflow
Title: ACMG/AMP Evidence Combination Logic
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in ACMG/AMP-CDx Research | Example/Catalog |
|---|---|---|
| Reference DNA Standards | Validate NGS panel sensitivity/specificity for known P/LP/VUS variants. Essential for CDx analytical validation. | Seraseq FFPE Tumor Mutation DNA, Horizon Discovery Multiplex I cfDNA Reference. |
| ACMG/AMP Rule-Based Software | Automates variant classification by applying evidence codes, reducing manual review time for large datasets. | Franklin by Genoox, VarSome Clinical, Fabric Genomics. |
| Clinical-Grade NGS Panels | Targeted sequencing kits designed for consistent coverage of cancer genes with validated performance for CDx development. | Illumina TruSight Oncology 500, Thermo Fisher Oncomine Precision Assay. |
| In Silico Prediction Tools | Provide computational evidence codes (PP3, BP4). Critical for initial variant assessment. | SIFT, PolyPhen-2, CADD, REVEL (Integrated in VEP, ANNOVAR). |
| Population Frequency Databases | Source for allele frequency evidence (PM2, BS1, BA1). Must be large and disease-appropriate. | gnomAD (non-cancer subset), Bravo, 1000 Genomes. |
| Disease-Specific Variant Databases | Curated repositories providing disease context and evidence (PM1, PS4, PP1). | ClinGen Expert Panels, ClinVar, COSMIC, OncoKB. |
| Cell Line Engineering Kits | Create isogenic models with specific VUS to generate functional data (PS3/BS3) for classification. | CRISPR-Cas9 Gene Editing Systems (e.g., Synthego). |
| CDx Algorithm Development Platform | Environment to build, lock, and validate the automated classification algorithm for regulatory submission. | DNAnexus, Seven Bridges, Google Cloud Life Sciences. |
1. Application Notes: Integrating Long-Read Sequencing Data into ACMG AMP Classification
Long-read sequencing (LRS) technologies from PacBio (HiFi) and Oxford Nanopore Technologies (ONT) are generating novel data types that challenge and enrich the traditional variant classification framework. These Application Notes outline their impact on specific ACMG/AMP criteria and provide a framework for their systematic incorporation.
Table 1: Impact of Long-Read Sequencing Data on ACMG/AMP Criteria
| ACMG/AMP Criterion | Traditional Data Source | Long-Read Sequencing Enhancement | Proposed Updated Application |
|---|---|---|---|
| PVS1 (Null variant) | Short-read RNA-seq, Sanger | Phased, full-length transcript sequencing. | Direct detection of allelic nonsense-mediated decay (NMD) or exon-skipping in cis. Resolves complex locus architectures. |
| PM3 (in trans for recessive) | Familial testing, haplotype analysis | Phasing over >100 kb distances. | Definitive determination of phase for compound heterozygotes in cis or trans without parental samples, especially in high-homology regions. |
| PM4 (Protein length change) | Short-read indel calling | Accurate resolution of repetitive/low-complexity regions. | Precise characterization of in-frame indels in tandem repeats (e.g., exon 20 dup in EGFR) previously misaligned. |
| PP3/BP4 (Computational evidence) | Short-read based predictors | Detection of cryptic splice variants, deep intronic, and structural variants. | Integration of LRS-derived splice effect predictions and in silico impact on non-coding elements from native DNA/RNA molecules. |
| BA1/BS2 (Allele frequency) | gnomAD (short-read) | More accurate allele frequency for complex variants. | Use of LRS-population resources (e.g., 1000 Genomes LR) to re-assess frequency of previously "uncallable" variants. |
2. Detailed Protocols
Protocol 2.1: Phasing for Compound Heterozygosity Determination (PM3)
Objective: To definitively determine the phase of two candidate variants in a recessive disorder gene using LRS without parental samples.
Materials:
- Genomic DNA (HMW recommended, >50 kb).
- PacBio Sequel II/IIe system with SMRTbell prep kit 3.0 or Oxford Nanopore PromethION with Ligation Sequencing Kit (SQK-LSK114).
- Target gene capture probes (e.g., Twist Bioscience) or whole-genome approach.
Methodology:
- Library Preparation & Sequencing: Prepare SMRTbell or ONT ligation libraries per manufacturer protocols, optionally enriched for the target gene/region. Sequence to achieve a minimum of 20x continuous coverage across the entire gene locus.
- Variant Calling & Phasing: Align reads to GRCh38 using minimap2. Call variants (small and structural) with tools like DeepVariant (PacBio) or Clair3 (ONT). Perform de novo phasing using the read-aware phaser WhatsHap or the integrated HiFi phased assembly pipeline (hifiasm).
- Analysis: Extract all reads spanning both variant positions. Compute the phase block length. If both variants reside on the same contiguous haplotype (in cis), they do not fulfill PM3. If they are consistently observed on separate haplotypes across multiple spanning reads (in trans), PM3 can be applied.
Protocol 2.2: Direct RNA Splicing Analysis for PVS1/PP3 Support
Objective: To characterize the splice-altering effect of a non-coding variant using native RNA long reads.
Materials:
- Patient and control cell line RNA (RIN > 8.5).
- Oxford Nanopore Direct RNA Sequencing Kit (SQK-RNA004) or PacBio Iso-Seq protocol reagents.
- Poly(A) mRNA selection beads.
Methodology:
- Library Preparation: For ONT, ligate sequencing adapter directly to 50-100 ng of poly(A)+ RNA. For PacBio, synthesize full-length cDNA, amplify, and prepare SMRTbell libraries.
- Sequencing & Alignment: Sequence to achieve ~5-10 million reads per sample. Align reads to the transcriptome/genome using minimap2 with splice-aware settings (
-ax splice). - Splice Isoform Analysis: Use tools like FLAIR (ONT) or Iso-Seq3 (PacBio) to collapse reads into non-redundant transcript models. Quantify the proportion of aberrant transcripts (exon skipping, intron retention, cryptic splice site usage) in patient vs. control. A >80% shift towards a truncating isoform provides strong evidence for PVS1; partial effects inform PP3/BP4 weighting.
3. Diagrams
Title: Long-Read Data Integration into ACMG Workflow
4. The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Reagents for Long-Read Variant Characterization
| Item | Supplier Examples | Function in Experiment |
|---|---|---|
| High Molecular Weight (HMW) DNA Isolation Kit | PacBio (MagiPrep), Qiagen (Genomic-tip), Circulomics (Nanobind) | Preserves ultra-long DNA fragments (>50 kb) critical for generating continuous, phaseable reads across large genomic regions. |
| Cas9-based Enrichment Kit (e.g., No-Amp) | PacBio, Twist Bioscience | Enriches for specific target genes/loci from complex genomes, increasing on-target LRS coverage cost-effectively for focused studies. |
| Direct RNA Sequencing Kit | Oxford Nanopore (SQK-RNA004) | Sequences native RNA molecules directly, enabling detection of base modifications and accurate quantification of full-length splice isoforms without cDNA bias. |
| HiFi SMRTbell Prep Kit 3.0 | PacBio | Prepares circularized, SMRTbell template libraries for PacBio Sequel IIe/Revio systems, generating highly accurate long reads (HiFi reads, Q > 30). |
| Ligation Sequencing Kit (V14) | Oxford Nanopore (SQK-LSK114) | Prepares DNA libraries for Nanopore sequencing by ligating sequencing adapters, optimized for high yield and duplex (high-accuracy) read recovery. |
| Reference Genome (T2T-CHM13) | Genome Reference Consortium | A complete, telomere-to-telomere reference assembly that resolves gaps and complex regions in GRCh38, essential for accurate LRS read alignment in previously problematic areas. |
Conclusion
The ACMG-AMP variant classification framework provides an indispensable, standardized lexicon for genomic interpretation, forming the bedrock of reproducible research and targeted drug development. Mastering its foundational principles, meticulous application, and nuanced troubleshooting is crucial for accurately translating genetic findings into biological insights and therapeutic hypotheses. As genomic data complexity grows, the continued evolution, refinement, and expert validation of these criteria will be paramount. Future directions include greater integration of quantitative modeling, automation-assisted classification, and adaptation for emerging modalities like polygenic risk and gene-gene interactions, ensuring its central role in the next generation of precision medicine.