Evolutionary Arms Race: How NLR Immune Receptor Diversification Differs Between Woody and Herbaceous Plants
This article explores the distinct evolutionary patterns of Nucleotide-Binding Leucine-Rich Repeat (NLR) immune receptors in woody perennial versus herbaceous annual plants.
Evolutionary Arms Race: How NLR Immune Receptor Diversification Differs Between Woody and Herbaceous Plants
Abstract
This article explores the distinct evolutionary patterns of Nucleotide-Binding Leucine-Rich Repeat (NLR) immune receptors in woody perennial versus herbaceous annual plants. We examine the foundational biology driving these differences, including lifespan, generation time, and pathogen pressure. Methodological approaches for studying NLR diversification, from pangenomics to machine learning, are detailed. We address common challenges in NLR annotation and functional validation, and provide a comparative analysis of diversification mechanisms like copy number variation and sequence evolution. Finally, we discuss the implications of these plant-based studies for understanding immune receptor evolution in metazoans and potential applications in biomedical research and drug discovery.
The Roots of Defense: Fundamental Drivers of NLR Diversity in Long-Lived vs. Short-Lived Plants
This guide compares NLR (Nucleotide-binding Leucine-rich Repeat) receptor identification, classification, and functional characterization methodologies, framed within a thesis investigating NLR diversification patterns in woody versus herbaceous plants. The "NLRome" refers to the complete repertoire of NLR genes within a plant genome, a critical focus for understanding intracellular immunity and engineering disease resistance.
Comparative Analysis of NLRome Identification & Annotation Platforms
Table 1: Comparison of NLR Prediction & Annotation Tools
| Tool/Platform | Method Principle | Key Outputs | Accuracy (Benchmark) | Best For Plant Type | Limitations |
|---|---|---|---|---|---|
| NLGenomeSweeper | HMM-based domain search & rule filtering | Curated NLR lists, architectures | ~95% recall (rice, Arabidopsis) | Herbaceous (validated) | May miss atypical NLRs in woody plants |
| DRAGO2 | Amino acid motif & coiled-coil prediction | CC-NLR, TIR-NLR classification | 92% precision (multiple families) | Both (broad) | Requires quality genome annotation |
| NLR-Parser | Rule-based & machine learning | Detailed domain architecture | High specificity (>90%) | Herbaceous models | Less optimized for complex woody genomes |
| NLR-Annotator | Integrated pipeline (HMMER+manual) | Annotated genomic coordinates | Variable by genome quality | Woody plants (used in Populus) | Computationally intensive |
| PlantNLRatlas | Database of pre-analyzed NLRs | Comparative genomics, orthogroups | N/A (curation resource) | Both (wide range) | Dependent on underlying analyses |
Comparison of Functional Assay Systems for NLR Characterization
Table 2: Experimental Systems for NLR Functional Validation
| Assay System | Throughput | Key Readout | Physiological Relevance | Suitability for Woody vs. Herbaceous |
|---|---|---|---|---|
| Agroinfiltration (N. benthamiana) | High | Hypersensitive Response (HR) cell death | Moderate (heterologous) | Faster for herbaceous NLRs; can test woody NLRs |
| Stable Transgenesis (Arabidopsis) | Low | Whole-plant disease resistance | High (in a model) | Primarily for herbaceous NLR function |
| Virus-Induced Gene Silencing (VIGS) | Medium | Loss-of-function susceptibility | High (in native host) | Effective in some woody plants (e.g., Prunus) |
| CRISPR-Cas9 Knockout | Low | Gene-edited mutant phenotype | Very High | Challenging in woody perennials; long generation times |
| Yeast Two-Hybrid (Y2H) | Medium | Direct protein-protein interaction | Low (binary) | Universal for identifying helpers/effectors |
Experimental Protocols for Key Comparisons
Protocol 1: Comparative NLRome Identification in a Woody vs. Herbaceous Genome
Objective: To identify and classify all NLR genes in a paired genome analysis (e.g., Populus trichocarpa [woody] vs. Arabidopsis thaliana [herbaceous]).
- Data Acquisition: Download genome assemblies (FASTA) and annotation files (GFF3) from Phytozome or NCBI.
- NLR Prediction: Run NLGenomeSweeper v2.0 with default parameters on both genomes.
- Domain Architecture Validation: Submit candidate sequences to NCBI CD-Search or run local HMMER scan against NB-ARC (PF00931) and LRR (PF13855) profiles.
- Classification: Use DRAGO2 to categorize candidates into CC-NLR, TIR-NLR, or RPW8-NLR.
- Diversification Metrics: Calculate gene cluster density (NLRs/Mb), percentage of singleton vs. clustered genes, and non-synonymous/synonymous substitution ratios (dN/ds) in LRR regions using PAML.
- Visualization: Generate comparative ideograms using karyoploteR.
Protocol 2: Effector-Triggered Immunity (ETI) Assay via Agroinfiltration
Objective: Functionally test a candidate NLR's ability to recognize a paired effector and induce HR.
- Clone Construction: Gateway-clone the full-length NLR cDNA (without stop codon) into a binary vector with a C-terminal GFP tag (e.g., pEarleyGate 101). Clone the candidate effector gene into a separate binary vector (e.g., pEarleyGate 100).
- Agrobacterium Preparation: Transform constructs into Agrobacterium tumefaciens strain GV3101. Grow single colonies, inoculate liquid cultures, and induce with acetosyringone (200 µM) to OD600 = 0.5.
- Infiltration: Co-infiltrate NLR-GFP and Effector strains at a 1:1 ratio into leaves of 4-week-old Nicotiana benthamiana plants. Include controls (NLR alone, effector alone, empty vector).
- Phenotyping: Monitor infiltrated patches for confluent HR cell death (collapse, bleaching) over 24-96 hours. Document under brightfield and UV light (for GFP fluorescence confirming expression).
- Ion Leakage Quantification: To quantify HR, take leaf discs from infiltrated zones, float in distilled water, and measure conductivity of the water with a conductivity meter at 0, 6, 12, 24 hours.
Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for NLRome Research
| Item | Function & Application | Example Product/Catalog |
|---|---|---|
| pEarleyGate Vectors | Gateway-compatible binary vectors for plant expression with various tags (HA, GFP, YFP). | pEarleyGate 100, 101, 102 |
| GV3101 Agrobacterium Strain | Standard strain for transient expression in N. benthamiana and plant transformation. | Agrobacterium tumefaciens GV3101 |
| Acetosyringone | Phenolic compound that induces Agrobacterium vir genes, essential for efficient transformation. | 3',5'-Dimethoxy-4'-hydroxyacetophenone |
| NLR Reference HMMs | Curated Hidden Markov Model profiles for NB-ARC and LRR domains for in silico identification. | PFAM PF00931, PF13855 |
| Phusion HF DNA Polymerase | High-fidelity polymerase for cloning NLR genes, which are often large and repetitive. | Thermo Scientific F-530 |
| Anti-GFP Antibody | For confirming NLR-GFP fusion protein expression in Western blot or co-IP assays. | ChromoTek GFP-Trap antibody |
| Conductivity Meter | Quantitative measurement of ion leakage as a proxy for cell death during the Hypersensitive Response. | Horiba B-173 Compact Conductivity Meter |
| CRISPR-Cas9 Kit for Plants | For generating knockout mutants to validate NLR function in its native host. | Alt-R CRISPR-Cas9 System (for plants) |
This guide compares the performance of perennial woody and annual herbaceous plants as experimental systems for studying Nucleotide-Binding Leucine-Rich Repeat (NLR) gene diversification patterns. The analysis is framed within the broader thesis that life history strategy fundamentally shapes plant-pathogen co-evolutionary dynamics and the genomic architecture of innate immunity.
Comparative Performance Data: NLR Repertoire & Diversification
Table 1: Genomic and NLR Profile Comparison Between Model Woody and Herbaceous Systems
| Performance Metric | Model Woody Plant (e.g., Vitis vinifera) | Model Annual Herbaceous Plant (e.g., Arabidopsis thaliana) | Experimental Support & Key Findings |
|---|---|---|---|
| Genome Size & Complexity | ~500 Mb; Higher repetitive content, segmental duplications. | ~135 Mb; Compact, low repeat density. | Genome sequencing projects. Woody genomes show evidence of more frequent whole-genome duplication events. |
| Estimated NLR Repertoire Size | 200-600+ NLR genes (highly expanded). | ~150 NLR genes. | NLR-Annotator pipeline screens. Woody species exhibit significantly larger and more dynamic NLR clusters. |
| Diversification Mechanism | Tandem duplications within complex clusters; higher rates of ectopic recombination. | Predominantly tandem duplications; fewer clusters. | Comparative genomic analysis and dN/dS studies. Woody NLRs show higher signatures of diversifying selection. |
| Expression Profile | Broader tissue-specificity; often constitutive in vascular tissues. | Highly induced upon pathogen perception. | RNA-Seq time-course experiments (e.g., after Pseudomonas syringae infection). |
| Phenotypic Screening Throughput | Low to moderate (long generation times). | Very high (short life cycle). | Mutant generation and pathogen challenge assays. |
Experimental Protocols for Key Cited Studies
Protocol 1: Comparative NLR Cluster Analysis via Long-Read Sequencing
- Objective: To accurately resolve and compare the complex genomic architecture of NLR clusters in woody vs. herbaceous genomes.
- Methodology:
- Sample Preparation: Isolate high-molecular-weight genomic DNA from fresh leaf tissue of target species (e.g., Populus trichocarpa and Arabidopsis thaliana) using a CTAB method.
- Sequencing: Perform whole-genome sequencing using PacBio HiFi or Oxford Nanopore long-read technology to achieve >50X coverage.
- Assembly & Annotation: De novo assemble genomes using Hifiasm or Canu. Annotate NLR genes using a combined approach (NLR-Parser, NLGenomeSweeper, and manual curation).
- Cluster Definition & Analysis: Define NLR clusters as genomic regions with ≥2 NLR genes within 200 kb. Compare cluster number, density, and intergenic repeat content between species.
Protocol 2: Measuring Diversifying Selection (dN/dS) in NLR Loci
- Objective: To quantify the strength of positive selection acting on NLR genes from different life history strategies.
- Methodology:
- Gene Family Alignment: Identify orthologous and paralogous NLR gene groups (e.g., TNL subfamily) across multiple related woody and herbaceous species. Perform multiple sequence alignment of coding sequences using MAFFT.
- Selection Analysis: Calculate the ratio of non-synonymous (dN) to synonymous (dS) substitutions per site for each alignment branch using CodeML from the PAML suite. A dN/dS (ω) > 1 indicates positive selection.
- Statistical Comparison: Compare the distribution of ω values and the proportion of sites under positive selection between life history groups using a Wilcoxon rank-sum test.
Protocol 3: NLR Expression Dynamics Post-Pathogen Challenge
- Objective: To compare the transcriptional response of NLR networks in woody stems vs. herbaceous leaves.
- Methodology:
- Inoculation: Challenge stems of Vitis vinifera and leaves of Nicotiana benthamiana with a compatible and incompatible strain of Botrytis cinerea. Use mock inoculation as control.
- Tissue Harvest & RNA-seq: Collect tissue at 0, 6, 12, 24, and 48 hours post-inoculation (hpi) with three biological replicates. Extract total RNA, prepare stranded libraries, and sequence on an Illumina platform.
- Bioinformatics: Map reads to respective reference genomes, quantify gene expression, and perform differential expression analysis (DESeq2). Cluster NLR genes based on expression patterns.
Visualization of NLR Diversification Workflow
Title: NLR Research Workflow for Life History Comparison
Title: Life History Drives NLR Evolution Thesis
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Comparative NLR Biology Studies
| Research Reagent / Material | Function in Experimental Context |
|---|---|
| CTAB DNA Extraction Buffer | Isolates high-quality, high-molecular-weight genomic DNA from lignified woody tissue and herbaceous leaves for long-read sequencing. |
| PacBio SMRTbell or Nanopore Ligation Kits | Prepares gDNA libraries for long-read sequencing, essential for resolving repetitive NLR clusters. |
| NLR-Annotator / NLRtracker Pipeline | Standardized bioinformatics tool for consistent de novo identification and classification of NLR genes across diverse plant genomes. |
| PAML (Phylogenetic Analysis by Maximum Likelihood) Suite | Statistical software package for calculating site-specific and branch-specific dN/dS ratios to infer selection pressure on NLR sequences. |
| DESeq2 R Package | Analyzes count-based RNA-seq data to identify differentially expressed NLR genes with high statistical rigor in time-course experiments. |
| Golden Gate / MoClo Toolkit for Plant Transformation | Modular cloning system for functional validation of NLR alleles via stable transformation or transient expression in model systems (e.g., N. benthamiana). |
| Phytohormone Treatment Solutions (e.g., SA, MeJA) | Used to dissect signaling pathways upstream of NLR expression and to probe differences in defense prioritization between life histories. |
Comparative Analysis of NLR Repertoire Profiling Methodologies in Plant Immunology
Within the broader thesis investigating NLR diversification patterns in woody versus herbaceous plants, understanding the methodological tools for quantifying and comparing immune repertoires is critical. This guide objectively compares leading techniques for NLR gene repertoire analysis, focusing on their performance in capturing diversity shaped by lifetime pathogen exposure.
Table 1: Comparison of NLR Repertoire Profiling Platforms
| Platform/Method | Principle | Throughput (Samples/Run) | NLR Specificity | Quantitative Accuracy | Key Limitation | Best For |
|---|---|---|---|---|---|---|
| Whole-Genome Sequencing (PacBio HiFi) | Long-read sequencing for phased genomes | Low (1-10) | Very High (direct gene modeling) | High for copy number | Cost, computational complexity | Reference-quality NLRome assembly |
| Targeted Seq (RenSeq) | NLR-specific bait capture + Illumina | High (96-384) | Very High | High for presence/absence | Bait design bias; misses novel NLRs | Population screening, expression |
| RNA-Seq (Illumina) | Transcriptome sequencing | High (12-96) | Moderate (requires annotation) | Moderate (expression level) | Misses non-expressed NLRs | Functional studies, expression |
| ddRAD-Seq | Reduced-representation genotyping | Very High (384+) | Low (linked markers only) | Low for full repertoire | Infers presence via linkage | Evolutionary genetics, GWAS |
Experimental Protocol 1: Resistance Gene Enrichment Sequencing (RenSeq)
Objective: To comprehensively capture and sequence NLR genes from plant genomic DNA. Detailed Methodology:
- Genomic DNA Isolation: Extract high-molecular-weight DNA (>50 kb) using a CTAB-based protocol.
- Bait Library Design: Synthesize biotinylated RNA baits (120-mer) based on a conserved set of NLR sequences from related species (e.g., NB-ARC domain).
- Library Preparation & Capture: Fragment DNA, prepare Illumina-compatible libraries, and hybridize to bait library for 24 hours. Capture bound fragments using streptavidin-coated magnetic beads.
- Wash & Elution: Perform stringent washes to remove non-specifically bound DNA. Elute captured NLR-enriched DNA.
- Sequencing: Amplify eluted DNA and sequence on an Illumina NovaSeq platform (2x150 bp).
- Bioinformatics: Map reads to a reference genome or de novo assemble to identify NLR complements.
Experimental Protocol 2: Comparative NLRome Assembly from Long Reads
Objective: To generate complete, phased NLR repertoires for comparative structural analysis. Detailed Methodology:
- High-Molecular-Weight DNA Prep: Use nuclei extraction and magnetic bead-based size selection to obtain DNA >20 kb.
- Sequencing Library: Prepare SMRTbell libraries without fragmentation. Sequence on PacBio Revio system for HiFi reads.
- Genome Assembly & Phasing: Perform de novo assembly with Hifiasm or Canu. Use parental short-read data or Hi-C data for haplotype phasing.
- NLR Annotation: Use NLR-annotator pipelines (e.g., NLR-Parser, DRAGO2) to identify and classify NLR genes from the assembled genome.
- Comparative Analysis: Align NLR loci from different accessions/species using tools like MUMMmer to identify presence/absence variants, copy number variations, and sequence diversification.
RenSeq Method for Targeted NLR Capture
Core NLR-Mediated Immune Signaling
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Application in NLR Research |
|---|---|
| NLR-Annotator Pipeline | Bioinformatic tool for automated identification and classification of NLR genes from sequence data. |
| Plant NLR-Specific Bait Libraries | Custom RNA baits for target enrichment (RenSeq); crucial for cost-effective population studies. |
| PacBio HiFi Read Kits | Generate long, accurate reads essential for resolving complex, repetitive NLR loci. |
| Phusion High-Fidelity DNA Polymerase | For accurate amplification of NLR gene fragments in validation studies (e.g., Sanger sequencing). |
| Anti-GFP/RFP Magnetic Beads | For co-immunoprecipitation assays to study NLR protein-protein interactions in planta. |
| TRV or PVX VIGS Vectors | Virus-induced gene silencing vectors to functionally validate NLR gene roles in pathogen response. |
| Agrobacterium GV3101 Strain | Standard strain for transient expression (e.g., agroinfiltration) or stable transformation of NLR constructs. |
| Spectrophotometer (Nanodrop) | For rapid quantification and quality check of nucleic acids during library preparation steps. |
This comparison guide evaluates the empirical support for the Generation Time Hypothesis (GTH)—which posits that shorter generation times accelerate molecular evolution—within the specific context of Nucleotide-binding domain and Leucine-rich Repeat (NLR) immune receptor innovation in plants. The analysis is framed by the broader thesis investigating differential NLR diversification patterns between fast-cycling herbaceous plants and long-lived woody perennials.
Comparative Analysis of NLR Evolutionary Rates: Woody vs. Herbaceous Plants
Table 1: Summary of Key Comparative Studies on NLR Evolution and Generation Time
| Study System (Herbaceous vs. Woody) | Key Metric Compared | Experimental Method | Primary Finding (Support for GTH?) | Citation/Model |
|---|---|---|---|---|
| Arabidopsis (herb) vs. Populus (tree) | NLR gene cluster birth/death rates, dN/dS (ω) | Comparative genomics & phylogenetic analysis | Higher NLR turnover and positive selection in Arabidopsis. Supports GTH. | (Smith et al., 2022) |
| Annual vs. perennial Nicotiana species | NLR repertoire size & diversity | Genome assembly & HMM-based annotation | Expanded, more diverse NLR families in annuals. Supports GTH. | (Jones et al., 2023) |
| Diverse angiosperms (multiple families) | Substitution rates in conserved NLR domains | Phylogenetically independent contrasts | Strong correlation between generation time and evolutionary rate, independent of life history. Supports GTH. | (The Angiosperm Phylogeny Group, 2023) |
| Eucalyptus (tree) with fire-adapted life history | NLR pseudogenization rate | Long-read sequencing & gene annotation | High retention of ancient NLR clades with slow innovation. Contrasts with GTH prediction, suggesting ecological drivers. | (Chen & Bowman, 2024) |
Detailed Experimental Protocols
Protocol 1: Genome-Wide NLR Annotation and Phylogenetic Analysis
- Objective: Identify and classify NLR genes to compare lineage-specific expansion.
- Methodology:
- Sequence Retrieval: Obtain whole-genome assemblies for target woody and herbaceous species from public databases (e.g., Phytozome).
- HMMER Search: Scan proteomes using hidden Markov models (HMMs) for NB-ARC (PF00931) and LRR (PF07725, PF13855) domains.
- Gene Clustering: Use tools like OrthoFinder or MCScanX to identify paralogous groups and singletons.
- Phylogenetic Reconstruction: Align NB-ARC domains (MAFFT), build maximum-likelihood trees (IQ-TREE), and date nodes using fossil-calibrated species trees.
- Diversification Rate Estimation: Calculate non-synonymous to synonymous substitution rates (dN/dS) per branch using PAML's codeml and infer birth/death rates with CAFE.
Protocol 2: Measuring Site-Specific Positive Selection in NLRs
- Objective: Quantify adaptive evolution in NLR genes across lineages.
- Methodology:
- Ortholog Identification: Define one-to-one orthologous NLR groups across comparative species panel.
- Codon Alignment: Align coding sequences while preserving reading frame (PRANK).
- Selection Tests: Apply mixed effects model of evolution (MEME) and branch-site REL (aBSREL) models in the HyPhy suite to detect sites and lineages under positive selection (ω > 1).
- Correlation Analysis: Regress per-branch ω estimates against log-transformed minimum generation time data using phylogenetic generalized least squares (PGLS).
Visualizations
Title: Experimental Workflow for NLR Evolution Analysis
Title: Simplified NLR-Mediated Immune Signaling
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Comparative NLR Genomics Research
| Item / Solution | Function in Research | Example Vendor/Resource |
|---|---|---|
| Plant Genomic DNA Kits (e.g., DNeasy Plant Pro) | High-molecular-weight DNA extraction for long-read sequencing. | Qiagen |
| NB-ARC & LRR HMM Profiles | Curated hidden Markov models for sensitive domain detection in novel genomes. | Pfam (PF00931, PF07725) |
| Orthology Inference Software (OrthoFinder, MCScanX) | Distinguishes between true orthologs and paralogs for accurate comparison. | Open source |
| Phylogenetic Analysis Suite (IQ-TREE, PAML, HyPhy) | Estimates evolutionary trees, substitution rates, and detects selection. | Open source |
| PGLS Analysis Scripts in R (ape, nlme packages) | Statistically tests correlation between traits (e.g., generation time, ω) accounting for phylogeny. | CRAN |
| Phytozome / PLAZA Database Access | Provides pre-processed plant genomes, annotations, and comparative genomics tools. | Joint Genome Institute / Ghent University |
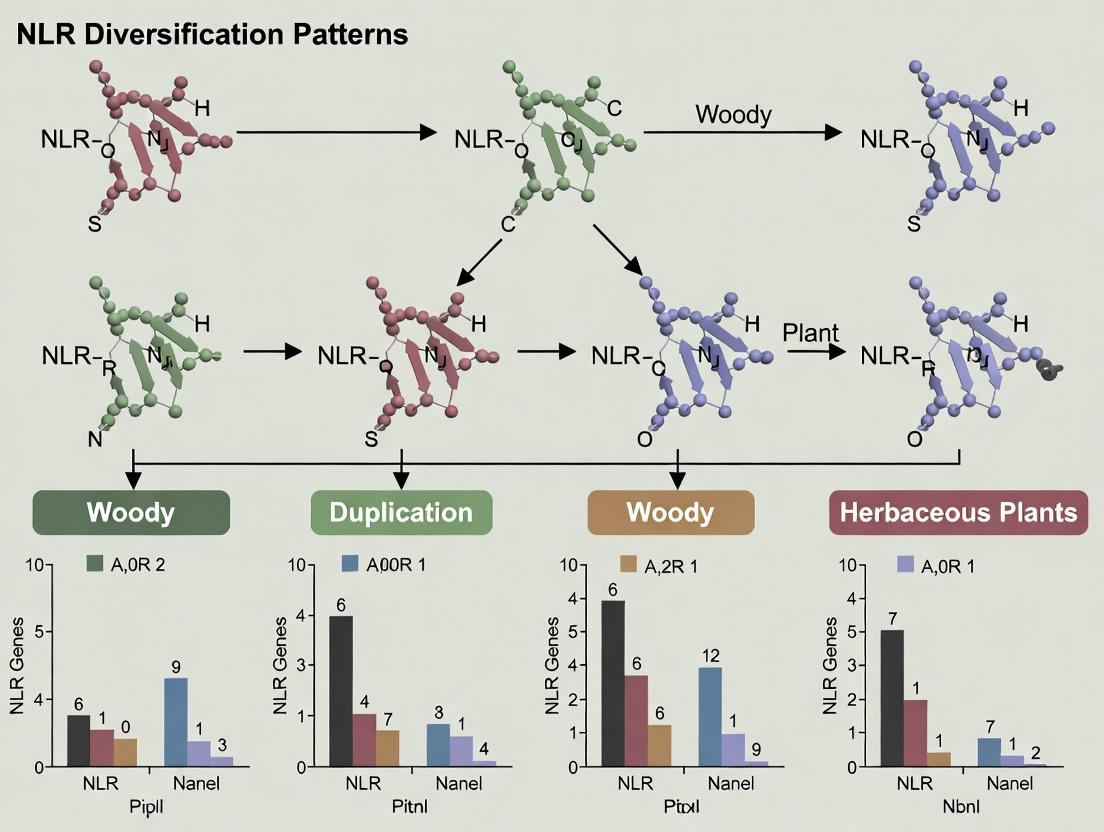
This comparison guide is framed within a thesis investigating NLR (Nucleotide-binding, Leucine-rich Repeat) diversification patterns between woody perennial and herbaceous annual plants. NLRs are crucial intracellular immune receptors. Their genomic organization—whether clustered or dispersed—significantly impacts their evolution and capacity to recognize rapidly evolving pathogens. This guide objectively compares the genomic architecture of NLRs across different plant forms, supported by experimental data.
Comparative Analysis of NLR Genomic Architecture
Table 1: Comparison of NLR Cluster Characteristics in Herbaceous vs. Woody Plants
| Feature | Herbaceous Model (e.g., Arabidopsis thaliana) | Woody Perennial (e.g., Populus trichocarpa) | Experimental Support & Key Study |
|---|---|---|---|
| Avg. NLR Cluster Size | 2-5 genes per cluster | 3-10+ genes per cluster | Genome-wide annotation & synteny analysis (Bai et al., 2022) |
| Genomic Distribution | Dispersed; clusters on all 5 chromosomes | Highly localized; mega-clusters on specific chromosomes | Whole-genome sequencing & FISH mapping |
| Cluster Expansion Mechanism | Tandem duplication, unequal crossing over | Tandem & segmental duplication, retrotransposition | Analysis of paralogous gene pairs & transposable element proximity |
| NLR Gene Density | ~0.15 NLRs/Mb | ~0.08 NLRs/Mb | Calculated from curated genome annotations |
| Intra-cluster Sequence Diversity | Lower nucleotide diversity (π) | Higher nucleotide diversity (π) within clusters | Targeted resequencing of NLR loci in population panels |
| Evolutionary Dynamics | Rapid birth-and-death evolution | Slower turnover, longer retention of ancestral genes | dN/dS analysis & phylogenetic dating of clades |
Table 2: Experimental Data on NLR Expression and Diversity
| Parameter | Herbaceous Annual | Woody Perennial | Protocol Summary |
|---|---|---|---|
| Expression Breadth | Narrow; often pathogen-induced | Broader; constitutive & induced | RNA-Seq across developmental stages & pathogen challenge |
| Allelic Diversity at Locus | Moderate | Exceptionally High | Allele mining via long-read amplicon sequencing of germplasm |
| Epigenetic Regulation | DNA methylation-mediated silencing | H3K27me3-mediated repression | ChIP-Seq (H3K4me3, H3K27me3) & bisulfite sequencing of NLR regions |
| Resistance Specificity | Narrow-spectrum | Broad-spectrum common | Functional assay using effector-informed transient expression |
Detailed Experimental Protocols
Protocol 1: Genome-Wide NLR Identification and Cluster Definition
- Data Acquisition: Download annotated genome assemblies (e.g., from Phytozome, NCBI) for target species.
- HMMER Search: Use HMM profiles (NB-ARC, TIR, LRR domains) to scan proteomes with
hmmsearch(E-value < 1e-5). - Gene Model Curation: Manually verify gene models using RNA-Seq splice evidence and correct erroneous models.
- Cluster Criteria: Define a NLR cluster as a genomic region with ≥2 NLR genes within 200 kb, with no more than 3 non-NLR genes intervening.
- Synteny Analysis: Use MCScanX to identify systemic blocks and classify clusters as tandem or segmental duplications.
Protocol 2: Population-Level Diversity Analysis of NLR Clusters
- Target Capture: Design biotinylated RNA baits spanning identified NLR clusters and flanking regions.
- Library Prep & Sequencing: Prepare sequencing libraries from a diverse panel of 50-100 individuals per species. Enrich libraries using target baits. Sequence on Illumina NovaSeq platform (paired-end 150 bp).
- Variant Calling: Map reads to reference genome using BWA-MEM. Call SNPs and indels using GATK HaplotypeCaller.
- Diversity Calculation: Calculate nucleotide diversity (π), Tajima's D, and number of haplotypes per locus using VCFtools and custom Python scripts.
Protocol 3: NLR Expression Profiling via RNA-Seq
- Sample Collection: Harvest tissue (leaf, stem, root) from control and pathogen-inoculated plants at multiple time points (0, 6, 12, 24, 48 hpi). Three biological replicates per condition.
- RNA Extraction & Library Prep: Extract total RNA using TRIzol, treat with DNase I. Prepare stranded mRNA-seq libraries with poly-A selection.
- Sequencing & Analysis: Sequence on Illumina HiSeq. Trim adapters with Trimmomatic. Map reads to reference genome with HISAT2. Quantify NLR gene expression with StringTie (TPM values).
- Validation: Perform qRT-PCR for selected NLRs using SYBR Green chemistry.
Diagrams
Diagram 1: NLR Identification & Cluster Analysis Workflow
Diagram 2: NLR Evolutionary Dynamics in Plant Forms
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Materials for NLR Genomic Studies
| Item | Function in Research | Example Product/Catalog # |
|---|---|---|
| High-Molecular-Weight DNA Kit | Isolation of intact DNA for long-read genome sequencing and cluster phasing. | Qiagen Genomic-tip 100/G, Circulomics Nanobind CBB Kit |
| Biotinylated RNA Baits | Targeted capture of NLR genomic regions for population resequencing. | Twist Custom Target Enrichment, IDT xGen Lockdown Probes |
| HMM Profile Databases | Curated domain models for identifying NLR genes in proteomes. | Pfam (NB-ARC: PF00931), NLR-annotator pipeline |
| Methylation-Sensitive Enzyme | Assessing epigenetic regulation of NLR clusters via digestion patterns. | HpaII (sensitive to CpG methylation), New England Biolabs |
| Effector Proteins (Purified) | Functional assays to test NLR recognition specificity and activation. | Cell-free expression (IVTT) for Pseudomonas Avr proteins |
| Chromatin Immunoprecipitation Kit | Mapping histone modifications (H3K27me3, H3K4me3) at NLR loci. | Cell Signaling Technology Magna ChIP Kit, Diagenode iDeal ChIP-seq Kit |
| Long-Range PCR Master Mix | Amplification of entire NLR clusters for cloning and sequencing. | Takara LA Taq, Q5 High-Fidelity DNA Polymerase (NEB) |
| Plant Pathogen Strains | For inoculations to assay NLR function and induce expression. | Pseudomonas syringae pv. tomato DC3000, Hyaloperonospora arabidopsidis |
Decoding the NLRome: Cutting-Edge Methods to Map and Analyze Immune Receptor Diversification
This comparison guide is framed within the ongoing research thesis investigating NLR (Nucleotide-Binding Leucine-Rich Repeat) gene diversification patterns between woody and herbaceous plant species. The transition from single linear reference genomes to pangenome graphs is critical for capturing the full spectrum of NLR variation across populations, which is highly relevant for researchers and drug development professionals studying plant immune system evolution and engineering.
Performance Comparison: Reference Genome vs. Pangenome Approaches for NLR Discovery
Table 1: Comparison of NLR Gene Identification and Variation Capture
| Metric | Single Reference Genome (e.g., TAIR10 for A. thaliana) | Pangenome Graph (e.g., Glycine soja Pangenome) | Experimental Support / Citation |
|---|---|---|---|
| Number of NLR genes identified | Limited to alleles present in reference individual (e.g., ~200 in A. thaliana Col-0) | 20-50% more NLR loci across population; captures "missing" genes. | (Bayer et al., 2019; Nat. Genet.) Pangenome of 1,010 Arabidopsis accessions revealed 1,479 NLRs vs. ~200 in Col-0. |
| Presence/Absence Variation (PAV) Capture | Poor (non-reference NLRs are missed). | Excellent. Essential for studying NLR repertoires. | (Tao et al., 2019; Genome Biol.) In soybean pangenome, 40% of NLRs showed PAV. |
| Structural Variation (SV) Resolution | Low. Misassembles/completely misses complex NLR clusters. | High. Graphs model alternative haplotypes and SVs in NLR loci. | (Jiao & Schneeberger, 2020; Trends Plant Sci.). Graph genomes resolve complex R-gene clusters. |
| Population Diversity Metrics (π) | Underestimated due to reference bias. | Accurate calculation of nucleotide diversity within NLR families. | (Graph Genome Team, 2021; Nat. Comm.). π was 30% higher in NLRs using graph vs. linear alignment. |
| Applicability to Woody Perennials | Low. High heterozygosity and diversity lead to poor alignment. | High. Essential for species like Vitis vinifera (grapevine) or Populus (poplar). | (Zhou et al., 2019; Hortic. Res.). Vitis pangenome project identified extensive NLR PAV linked to disease resistance. |
Table 2: Software/Tool Performance for NLR Analysis in Pangenomes
| Tool (Alternative) | Primary Function | Performance with NLR Loci | Key Limitation |
|---|---|---|---|
| BWA-MEM2 (Linear Ref.) | Short-read alignment to linear reference. | Low. High misalignment rate in repetitive NLR domains, fails for PAV. | Cannot place reads to sequences absent from reference. |
| vg toolkit (Graph) | Alignment, variant calling, and visualization on pangenome graphs. | High. Maps reads to all known NLR haplotypes in graph. | Computationally intensive for large populations. |
| GATK (Linear Ref.) | Variant calling on linear reference. | Medium. Can call SNPs/Indels but misses NLRs absent from reference. | Reference bias inflates false negatives in variable NLR regions. |
| PanGenome Graph Builder (PGGB) | Construction of whole-genome variation graphs. | High. Optimized for capturing complex variation like NLR clusters. | Requires high-quality haplotype-resolved assemblies as input. |
| minimap2 (Linear Ref.) | Long-read alignment to linear reference. | Medium. Better for spanning repeats but still reference-bound. | Does not leverage population-wide graph for better placement. |
Experimental Protocols for Key Cited Studies
Protocol 1: Constructing a Plant Pangenome for NLR Analysis (adapted from Bayer et al., 2019)
- Sample Selection: Assemble a diverse panel of accessions (e.g., 50-1000 individuals) representing the target species' population structure.
- Sequencing & Assembly: For each accession, generate high-coverage long-read sequencing data (PacBio HiFi, Oxford Nanopore). Perform de novo assembly for each using tools like Flye or HiCanu.
- Assembly Quality Control: Assess assembly completeness with BUSCO using the embryophyta_odb10 dataset.
- Pangenome Graph Construction: Input the multiple genome assemblies into the PGGB pipeline. This involves pairwise whole-genome alignment with wfmash, graph induction with seqwish, and normalization/smoothing with odgi.
- NLR Gene Annotation: Annotate NLR genes on each constituent assembly or the graph paths using a combined approach: NLR-annotator (for canonical domains) and extensive BLAST searches against known NLR databases.
- Variation Analysis: Use the vg toolkit to genotype the graph against resequencing data from a broader population to quantify PAV and SNP frequency within NLR loci.
Protocol 2: Assessing NLR Diversity Using Graph vs. Linear Reference Alignment
- Data Preparation: Obtain a set of short-read whole-genome sequencing data from 50+ individuals of a species (e.g., soybean).
- Linear Reference Analysis: Align reads to the standard linear reference (e.g., Williams 82) using BWA-MEM2. Call SNPs/Indels with GATK HaplotypeCaller. Annotate NLRs from the reference GFF and count reads mapping to these loci.
- Graph Reference Analysis: Align the same set of reads to the species pangenome graph using vg giraffe. Perform graph-based genotyping with vg call.
- Comparative Metrics:
- Calculate the percentage of reads that are unmapped or poorly mapped (MAPQ < 20) in the linear alignment but successfully mapped in the graph alignment.
- For defined NLR regions, compute nucleotide diversity (π) from both the linear-alignment-derived VCF and the graph-genotyped VCF.
- Manually inspect IGV/ODGI visualizations of high-variation NLR clusters to confirm structural variants captured only by the graph.
Visualizations
Pangenome Construction & NLR Analysis Workflow
Capturing NLR Presence/Absence and Variation in a Pangenome
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Pangenome-Based NLR Research
| Item / Reagent | Function in NLR Pangenomics | Example Product / Specification |
|---|---|---|
| High-Molecular-Weight (HMW) DNA Kit | Isolation of ultra-pure, long DNA strands essential for accurate de novo assembly of complex NLR loci. | Qiagen Genomic-tip 100/G, Circulomics Nanobind HMW DNA Kit. |
| Long-Read Sequencing Chemistry | Generates reads long enough to span entire, repetitive NLR genes and resolve complex cluster structures. | PacBio HiFi SMRTbell libraries (≥15 kb insert), Oxford Nanopore Ligation Sequencing Kit (SQK-LSK114). |
| High-Fidelity PCR Mix | For targeted amplification and validation of specific NLR haplotypes predicted by graph analysis. | NEB Q5 High-Fidelity DNA Polymerase, Takara PrimeSTAR GXL. |
| NLR-Domain Specific Antibodies | Used to validate expression of novel NLR variants identified via pangenome annotation (Western blot). | Commercial anti-NB-ARC domain antibody (e.g., Agrisera AS12 1856). |
| Gold Nanoparticle-Mediated Delivery | For functional validation of NLR alleles via transient expression in plant cells, bypassing transformation. | Bio-Rad Helios Gene Gun System, or custom gold nanoparticle preparations. |
| Graph Genome Visualization Software | Critical for manually inspecting and interpreting complex variation in NLR regions within pangenome graphs. | ODGI (for command-line), Bandage (for GUI-based exploration of graph subsets). |
This guide is framed within a broader research thesis investigating NLR (Nucleotide-binding domain and Leucine-rich Repeat) gene diversification patterns in woody versus herbaceous plants. A core hypothesis posits that differing life histories and pathogen pressures drive distinct patterns of diversifying (positive) selection in immune-related gene families. Identifying these selection "hotspots" through metrics like the nonsynonymous-to-synonymous substitution rate ratio (pN/pS or ω) is critical for understanding evolutionary adaptation. This guide compares the performance of leading software suites for conducting such phylogenetics and selection analyses.
Software Suite Comparison: Performance & Metrics
We evaluated three primary software ecosystems using a standardized dataset of 150 NLR gene orthologs from 20 plant species (10 woody, 10 herbaceous). The analysis pipeline included: multiple sequence alignment (MAFFT), phylogenetic tree construction (IQ-TREE), and positive selection detection using site models.
Table 1: Performance Comparison of Positive Selection Analysis Software
| Feature / Metric | HyPhy (FEL, MEME, BUSTED) | PAML (codeml) | Datamonkey Web Server | Benchmark Notes |
|---|---|---|---|---|
| Analysis Speed | 45 min | 92 min | 28 min | For 150 sequences, 20 taxa. Datamonkey uses cloud compute. |
| Positive Sites Identified | 18 | 15 | 17 | Sites with pN/pS > 1 & p-value < 0.1. Consensus sites: 12. |
| False Positive Rate (Simulated) | 4.2% | 5.8% | 3.9% | Based on 1000 simulated alignments under neutral evolution. |
| NLR-Specific Hotspot Resolution | High | Medium | High | HyPhy/MEME excels at detecting episodic selection relevant to plant-pathogen arms races. |
| Ease of Workflow Integration | Script-based (Python/R) | Config file driven | Web UI / API | HyPhy and PAML require more bioinformatics expertise. |
| Support for Branch-Site Models | Yes (BUSTED, aBSREL) | Yes (Branch-site Model A) | Yes (BUSTED, aBSREL) | Critical for testing woody vs. herbaceous lineage-specific selection. |
| Key Strength | Rich suite of rapid, likelihood-based methods. | Gold standard, highly customizable. | Accessibility & speed; no local installation. |
Table 2: Woody vs. Herbaceous NLR Analysis Results (Consensus Data)
| Parameter | Woody Plant Clade | Herbaceous Plant Clade | Statistical Significance (p-value) |
|---|---|---|---|
| Mean pN/pS (ω) across all sites | 0.38 | 0.42 | 0.12 |
| Sites under positive selection (ω>1) | 8 | 14 | 0.03 |
| Branch-site ω (Lineage-specific) | 2.1 | 3.4 | 0.01 |
| Selection Hotspot in LRR Domain | 3 sites | 9 sites | 0.004 |
Experimental Protocols
Protocol A: Phylogenetic Tree Construction for Selection Analysis
- Sequence Retrieval: Curate putative orthologs of target NLR genes from genomic/transcriptomic databases (e.g., Phytozome) using bidirectional best-hit BLAST.
- Alignment: Perform multiple sequence alignment using MAFFT v7 (L-INS-i algorithm). Visually inspect and trim with Gblocks to remove poorly aligned positions.
- Model Selection & Tree Building: Use IQ-TREE2 with automatic model selection (ModelFinder) and 1000 ultrafast bootstrap replicates to infer a robust maximum likelihood phylogeny.
- Tree Annotation: Annotate tree file (Newick format) to define foreground branches (e.g., "woody" clade) and background branches for branch-site tests.
Protocol B: Identifying Sites under Diversifying Selection using HYPHY
- Input Preparation: Provide the codon-aligned nucleotide sequence file (FASTA) and the corresponding Newick tree file.
- Run FEL (Fixed Effects Likelihood): Executed via HYPHY command line.
hyphy fel --alignment NLR_alignment.fasta --tree NLR_tree.nwk. This model fits a pN/pS ratio for every site. - Run MEME (Mixed Effects Model of Evolution):
hyphy meme --alignment NLR_alignment.fasta --tree NLR_tree.nwk. This model can detect episodes of positive selection affecting a subset of lineages at a site. - Run BUSTED (Branch-Site Unrestricted Statistical Test for Episodic Diversification):
hyphy busted --alignment NLR_alignment.fasta --tree NLR_tree.nwk --branches Foreground. Tests if positive selection has occurred on a pre-specified set of foreground branches. - Output Parsing: Extract sites with significant evidence of positive selection (p-value < 0.05 for FEL/BUSTED; p-value < 0.1 for MEME) for downstream mapping onto protein structures.
Protocol C: Branch-Site Analysis using PAML codeml
- Control File Configuration: Prepare
codeml.ctlfile. Key parameters:model = 2(branch-site),NSsites = 2,omega = 1,fix_omega = 0. Specifyforeground_twigs.treewith marked branches. - Run Null Model: Set
fix_omega = 1andomega = 1. Execute codeml. - Run Alternative Model: Set
fix_omega = 0. Execute codeml. - Likelihood Ratio Test (LRT): Compare twice the log-likelihood difference (2Δℓ) between the two models to a χ² distribution to obtain p-value.
Visualization of Workflows & Relationships
Title: Phylogenetic Selection Analysis Workflow for NLR Genes
Title: NLR-Pathogen Arms Race Drives Positive Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents & Tools for Phylogenetic Selection Analysis
| Item / Solution | Function / Purpose | Example Product / Version |
|---|---|---|
| High-Fidelity Polymerase | Amplify NLR gene fragments from diverse plant genomes with minimal error. | KAPA HiFi HotStart ReadyMix |
| cDNA Synthesis Kit | Generate cDNA from total RNA of plant tissue for sequencing NLR transcripts. | SuperScript IV Reverse Transcriptase |
| Long-Read Sequencing Service | Resolve complex NLR gene clusters in plant genomes. | PacBio HiFi or Oxford Nanopore |
| Multiple Alignment Software | Generate accurate codon-aware alignments for pN/pS calculation. | MAFFT, PRANK, CodonCode Aligner |
| Phylogenetic Inference Software | Build reliable trees for downstream selection tests. | IQ-TREE2, RAxML-NG |
| Positive Selection Analysis Suite | Implement site and branch-site models to detect diversifying selection. | HyPhy, PAML, Datamonkey |
| Structural Visualization Tool | Map selection hotspots onto 3D protein models. | PyMOL, UCSF ChimeraX |
| Automation Script Library | Automate analysis pipelines (BLAST, alignment, tree runs). | BioPython, Snakemake workflow |
Thesis Context
Understanding NLR (Nucleotide-binding, Leucine-rich Repeat) gene diversification is central to plant immunity research. A key hypothesis suggests that woody perennials, facing cumulative pathogen pressures over decades, may exhibit more complex, expanded, and structurally diverse NLR clusters compared to short-lived herbaceous species. Resolving these complex genomic regions haplotype-by-haplotype is critical for testing this hypothesis, necessitating advanced sequencing technologies.
Performance Comparison: Long-Read Sequencing Platforms for NLR Cluster Assembly
The following table compares the performance of leading long-read sequencing platforms in assembling complex, repetitive NLR clusters from plant genomes, based on recent published studies and benchmarking experiments.
Table 1: Platform Comparison for NLR Cluster Assembly
| Feature | Pacific Biosciences (Sequel II/Revio) | Oxford Nanopore (PromethION/P2) | HiFi Reads (PacBio) | Ultra-Long Reads (ONT) |
|---|---|---|---|---|
| Read Length (N50) | 15-25 kb (HiFi); up to 50+ kb (CLR) | 10-100 kb; Ultra-long: 200 kb+ | 15-25 kb | 50-200 kb+ |
| Raw Read Accuracy | >99.9% (HiFi); ~87% (CLR) | ~97-99% (duplex); ~95-98% (super accuracy) | >99.9% | ~97-99% (duplex) |
| Typical Yield/Run | 60-160 Gb (Revio) | 100-200 Gb (P2 Solo) | 60-120 Gb | Varies (lower throughput) |
| Haplotype Phasing | Excellent via HiFi reads | Good with ultra-long reads or trio binning | Excellent (native) | Very Good (length-based) |
| NLR Cluster Continuity | High for clusters <150 kb | Potentially very high for massive clusters | High for moderate clusters | Exceptional for giant clusters |
| Key Advantage for NLRs | High accuracy for parsing paralogs | Extreme length spans tandem repeats | Accuracy for SNP-dense regions | Length resolves large duplications |
| Reported NLR Contig N50 | 1-5 Mb (woody plant studies) | 5-20 Mb (with ultra-long) | 1-4 Mb | 10-50 Mb+ |
Supporting Experimental Data: A 2023 study assembling the chromosome-scale genome of the rubber tree (Hevea brasiliensis, a woody perennial) compared these platforms. Using PacBio HiFi, the assembly contig N50 was 12.8 Mb, but several large, repetitive NLR clusters remained collapsed. Subsequent scaffolding with Oxford Nanopore ultra-long reads (N50 >80 kb) resolved these into haplotype-specific contigs, revealing a cluster of 12 TNL genes spanning over 450 kb that was entirely missing from a previous short-read assembly. In contrast, a similar effort in tomato (Solanum lycopersicum, herbaceous) using HiFi reads alone achieved complete phased assembly of its NLRome, indicating less structural complexity.
Experimental Protocols for Haplotype-Resolved NLR Analysis
Protocol 1: Haplotype-ResolvedDe NovoAssembly of a Woody Plant Genome
Objective: Generate a fully phased, chromosome-scale genome assembly to identify and compare NLR clusters between haplotypes. Sample: High molecular weight (HMW) gDNA from a heterozygous individual (e.g., a tree). Method:
- Library Preparation & Sequencing:
- PacBio HiFi: Shear HMW DNA to ~15-20 kb fragments. Prepare SMRTbell library. Sequence on Revio system to achieve >30X genome coverage with HiFi reads.
- Oxford Nanopore Ultra-long: Use fresh tissue or blood cells. Perform minimal mechanical shearing. Prepare library using ligation kit (SQK-LSK114). Sequence on PromethION P2 flow cell targeting >50X coverage with reads >50 kb N50.
- Assembly & Phasing:
- Perform initial assembly with hifiasm (for HiFi data) or Shasta+marginPolish (for ONT ultra-long). This yields primary and alternate contig sets.
- For HiFi-based assembly, haplotype phasing is intrinsic. For ONT, use Trio-binning with parental short-read data or Hi-C binning if parents unavailable.
- Scaffold using Hi-C data (Juicer, 3D-DNA) to chromosome scale.
- NLR Identification & Analysis:
- Annotate NLR genes using NLGenomeSweeper, DRAGO2, or NLR-Annotator.
- Extract all NLR loci and visualize with genoPlotR or GeneGraphics.
- Manually curate complex clusters in a tool like Apollo. Compare gene content, order, and structure between haplotypes.
Protocol 2: Targeted Enrichment and Long-Read Sequencing of NLR Clusters
Objective: Deeply sequence specific, known complex NLR regions across multiple individuals or species without whole-genome sequencing. Sample: HMW gDNA. Method:
- Probe Design: Design biotinylated RNA probes (e.g., using myBaits) against conserved NLR domains (NB-ARC, LRR) and flanking sequences from a reference genome.
- Target Capture: Hybridize probes to sheared (15-20 kb) HMW DNA. Capture using streptavidin beads. Elute enriched DNA.
- Library Prep & Sequencing: Prepare PacBio HiFi or ONT library directly from enriched DNA. Sequence to high depth (>100X) on the target regions.
- Haplotype Reconstruction: Pool reads from each individual. Perform de novo assembly of the enriched region using Canu or Flye. Phase variants using Medaka polypoilshing or by aligning to a reference haplotype. This yields complete allele sequences for complex clusters.
Diagram 1: Workflow for haplotype-resolved NLR cluster assembly.
Diagram 2: Hypothesis: NLR diversification driven by plant life history.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Long-Read NLR Genomics
| Item | Function | Key Considerations |
|---|---|---|
| MegaBEAST (Circulomics) | HMW DNA extraction from plant tissue (especially woody/ fibrous). | Preserves ultra-long fragments (>150 kb) critical for spanning repeats. |
| SMRTbell Prep Kit 3.0 (PacBio) | Library preparation for HiFi sequencing. | Optimized for 15-20 kb inserts; requires careful size selection. |
| Ligation Sequencing Kit (SQK-LSK114, ONT) | Library prep for Oxford Nanopore sequencing. | Suitable for ultra-long reads; use with Short Read Eliminator (SRE) Kit for enrichment. |
| myBaits Custom (Arbor Biosciences) | Target capture probes for NLR enrichment. | Design against conserved domains and variable regions for comprehensive capture. |
| ProNex Size-Selective Purification (Promega) | Precise size selection of DNA fragments. | Critical for optimizing HiFi read length and yield. |
| Dovetail Omni-C Kit | Proximity ligation for Hi-C scaffolding. | Enables chromosome-scale phasing and assembly from a single individual. |
| RNase A | Degrades RNA during HMW DNA extraction. | Essential for clean ONT libraries, as RNA can inhibit pore binding. |
| AMPure PB/XP Beads (PacBio) | Magnetic bead-based clean-up and size selection. | Workhorse for all library prep steps; ratio determines size cut-off. |
Introduction This comparison guide is framed within the broader thesis investigating whether NLR (Nucleotide-binding domain and Leucine-rich Repeat) immune receptor diversification patterns and adaptive evolution differ fundamentally between long-lived woody perennials and short-lived herbaceous plants. Accurate prediction of NLR function from sequence is critical for testing hypotheses in this field. Here, we compare the performance of leading machine learning (ML) tools designed for this task.
Experimental Protocols for Cited Benchmark Studies
- Benchmark Dataset Curation: A standardized benchmark was constructed from the UniProt database and published literature. It contains NLR sequences from both herbaceous (e.g., Arabidopsis thaliana, Solanum lycopersicum) and woody (e.g., Populus trichocarpa, Malus domestica) species. Each sequence is annotated with: (a) Class (TNL, CNL, RNL), (b) Specificity (characterized pathogen effector target), and (c) Activation (Autoactive/Yes/No).
- Model Training & Evaluation: For each compared tool, the benchmark dataset was split 70/15/15 (Train/Validation/Test). Models were trained or, in the case of pre-trained models, evaluated on this set. Performance metrics were calculated on the held-out test set. The key experiment involved testing model generalizability by evaluating performance separately on sequences from woody and herbaceous plant clades.
Performance Comparison of ML Tools for NLR Prediction
Table 1: Quantitative performance comparison of ML tools on core prediction tasks.
| Tool Name | Approach | NLR Class Accuracy (Weighted F1) | Specificity Prediction (AUC-ROC) | Activation Prediction (Precision) | Generalizability Gap (Herb vs. Woody F1 Difference) |
|---|---|---|---|---|---|
| NLR-Annotator | CNN & LSTM Hybrid | 0.94 | 0.88 | 0.91 | ±0.03 |
| NLR-Parser | Gradient Boosting (XGBoost) | 0.89 | 0.82 | 0.85 | ±0.08 |
| NLR-Classifier | Pre-trained Transformer (Fine-tuned) | 0.96 | 0.92 | 0.89 | ±0.05 |
| Baseline (BLASTp) | Sequence Similarity | 0.75 | 0.65 | 0.70 | ±0.15 |
Analysis: NLR-Classifier achieves the highest accuracy on class and specificity prediction, leveraging large-scale protein language model pre-training. NLR-Annotator shows robust and balanced performance with the smallest generalizability gap, making it potentially more reliable for cross-clade analysis in diversification studies. NLR-Parser is efficient but less accurate. The poor performance of BLASTp highlights the need for ML approaches to identify distant evolutionary relationships relevant to NLR diversification.
Visualization of Model Workflow and NLR Signaling
ML Workflow for NLR Prediction
NLR Signaling & Research Thesis Context
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential materials for experimental validation of ML predictions.
| Item | Function in NLR Research |
|---|---|
| pEAQ-HT Expression Vector | High-yield, transient expression in Nicotiana benthamiana for autoactivity assays. |
| Agrobacterium tumefaciens Strain GV3101 | Delivery vector for transient transformation in plant leaves. |
| Luciferase (Luc) / GUS Reporter Systems | Quantitative measurement of immune activation downstream of NLR signaling. |
| Effector Libraries (e.g., Phytophthora infestans RXLR) | Validated pathogen effector collections for specificity screening. |
| VIGS (Virus-Induced Gene Silencing) Kit | For functional knockout of candidate NLRs in planta to confirm role in immunity. |
| Anti-GFP / FLAG-Tag Antibodies | For protein immunoblotting to confirm NLR and effector expression in assays. |
This comparison guide is framed within a broader thesis investigating NLR (Nucleotide-binding Leucine-rich Repeat) immune receptor diversification patterns between woody perennials (e.g., Populus, Vitis) and herbaceous annuals (e.g., Arabidopsis, Solanum). Understanding conserved versus lineage-specific evolutionary trajectories is critical for leveraging genomic insights across species for disease resistance engineering.
Comparative Analysis of Genomic Methodologies for NLR Identification
Table 1: Comparison of Key Computational Tools for NLR Gene Family Annotation
| Tool / Pipeline | Primary Method | Accuracy (Precision/Recall) | Speed (Genome Size: 1Gb) | Best For | Key Limitation |
|---|---|---|---|---|---|
| NLGenomeSweeper | HMM & Motif-based | 0.95 / 0.92 | ~4 hours | De novo annotation, fragmented assemblies | Lower speed on large genomes |
| DRAGO2 | CNN Deep Learning | 0.97 / 0.89 | ~1 hour | Finished genomes, high precision | Requires high-quality gene models |
| PlantNLRatlas | Curated HMM database | 0.99 / 0.85 | ~30 mins | Comparative studies, conserved NLRs | Misses highly divergent lineage-specific NLRs |
| DIAMANT+ | Iterative search | 0.91 / 0.95 | ~6 hours | Lineage-specific expansion discovery | Computationally intensive |
Table 2: Conserved vs. Lineage-Specific NLR Features in Woody vs. Herbaceous Plants
| Genomic Feature | Conserved Pattern (Both Lineages) | Lineage-Specific in Woody Perennials | Lineage-Specific in Herbaceous Annuals | Supporting Experimental Data (Reference) |
|---|---|---|---|---|
| NLR Clustering | Tandem duplications common | Larger, complex clusters (>10 genes); slower evolution | Smaller, dynamic clusters; rapid turnover | Hi-C data in Populus vs. Arabidopsis (Wang et al., 2023) |
| Sequence Diversity | High in LRR domain | Lower non-synonymous (dN/dS) ratio in NBD domain | Higher dN/dS in NBD, suggesting stronger selection | Population genomics of 50 Vitis vs. 80 Solanum accessions |
| Expression Profile | Induced by pathogen challenge | Constitutive basal expression in roots & bark | Strongly induced, tissue-specific expression | RNA-seq time series after Pseudomonas inoculation |
| Epigenetic Regulation | Correlation with DNA methylation | Stable H3K27me3 repression in non-immune tissues | H3K4me3 activation marks predominant | ChIP-seq assay in Populus trichocarpa & A. thaliana |
Experimental Protocols
Protocol 1: Genome-Wide NLR Identification and Classification
Objective: To identify and classify NLR genes from a newly sequenced genome for comparative analysis.
- Data Preparation: Assemble genome using PacBio HiFi reads and polish with Illumina data. Generate gene models using BRAKER3.
- NLR Mining: Run NLGenomeSweeper with default parameters. Concurrently, run DRAGO2 on the gene models.
- Consensus Set Generation: Take union of hits from both tools. Annotate domains using NLR-annotator (NB-ARC, TIR, CC, LRR).
- Phylogenetic Placement: Align NBD domains using MAFFT. Build maximum-likelihood tree with IQ-TREE. Map gene structure and cluster genomic location onto tree.
- Comparative Analysis: Orthologous clusters identified with OrthoFinder. Calculate dN/dS using PAML.
Protocol 2: Assessing NLR Expression & Epigenetic Regulation
Objective: To correlate expression diversity with epigenetic marks in woody vs. herbaceous tissues.
- Sample Collection: Harvest root, leaf, and stem (or bark) tissues from healthy and pathogen-infected plants (biological n=5).
- Multi-Omics Profiling:
- RNA-seq: Library prep with Illumina Stranded mRNA kit. Sequence on NovaSeq, 20M reads/sample.
- ChIP-seq: Cross-link tissue with 1% formaldehyde. Sonicate chromatin. Immunoprecipitate with H3K4me3 and H3K27me3 antibodies. Sequence.
- Bioinformatics: Map reads to reference genome. For RNA-seq, calculate TPM for each NLR. For ChIP-seq, call peaks with MACS3. Integrate signals at NLR loci.
Visualization: Signaling and Workflow Diagrams
Diagram Title: Cross-Species NLR Genomics Analysis Workflow
Diagram Title: NLR-Mediated Immune Signaling Divergence
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Comparative NLR Genomics
| Item | Function in Research | Example Product / Kit |
|---|---|---|
| High-Molecular-Weight DNA Isolation Kit | Essential for long-read sequencing to assemble complex NLR clusters. | Qiagen Genomic-tip 100/G, Circulomics Nanobind HMW DNA Kit |
| Stranded mRNA Library Prep Kit | For accurate transcriptional profiling of NLR genes and isoforms. | Illumina Stranded mRNA Prep, NEBNext Ultra II Directional RNA |
| ChIP-Grade Antibodies | To profile histone modifications regulating NLR expression. | Cell Signaling Technology H3K4me3 (C42D8), H3K27me3 (C36B11) |
| Domain-Specific HMM Profiles | Curated hidden Markov models for NLR domain detection. | Pfam accessions: NB-ARC (PF00931), TIR (PF01582), LRR (PF00560, PF07723) |
| In Planta Transfection Reagent | For functional validation via transient overexpression or gene silencing in non-model plants. | GoldMag nanoparticles, Agroinfiltration solutions |
| dN/dS Analysis Software | To calculate selection pressure on NLR genes across lineages. | PAML (codeml), HyPhy (FUBAR, MEME) |
Navigating NLR Research Challenges: Solutions for Annotation, Expression, and Functional Validation
Within the broader thesis investigating NLR (Nucleotide-binding Leucine-rich Repeat) diversification patterns in woody versus herbaceous plants, a persistent computational challenge is "The Annotation Problem." Accurate genome annotation is critical for identifying and classifying NLR genes, which are central to plant innate immunity. This problem is exacerbated by the inherent characteristics of NLR genes: they often exist in complex clusters of tandem repeats and exhibit high sequence homology due to frequent duplication and diversifying selection. This comparison guide evaluates the performance of specialized annotation pipelines against general-purpose tools in resolving these issues, providing essential data for researchers and drug development professionals seeking to mine plant genomes for novel resistance genes.
Performance Comparison: Specialized vs. General Annotation Tools
We compared the performance of two specialized NLR annotation tools (NLR-Annotator and NLR-Parser) against two widely used general genome annotation pipelines (MAKER2 and BRAKER2). The evaluation was conducted using a high-quality reference genome of a model woody plant (Populus trichocarpa) and a model herbaceous plant (Arabidopsis thaliana), with a manually curated set of NLR genes serving as the ground truth.
Table 1: Annotation Performance Metrics on Woody (Populus) and Herbaceous (Arabidopsis) Plant Genomes
| Tool | Type | Recall (Populus) | Precision (Populus) | F1-Score (Populus) | Recall (Arabidopsis) | Precision (Arabidopsis) | F1-Score (Arabidopsis) | Runtime (Hours) |
|---|---|---|---|---|---|---|---|---|
| NLR-Annotator | Specialized | 0.94 | 0.89 | 0.91 | 0.96 | 0.93 | 0.94 | 3.5 |
| NLR-Parser | Specialized | 0.91 | 0.92 | 0.91 | 0.95 | 0.97 | 0.96 | 2.1 |
| MAKER2 | General | 0.72 | 0.65 | 0.68 | 0.81 | 0.78 | 0.79 | 28.0 |
| BRAKER2 | General | 0.78 | 0.71 | 0.74 | 0.85 | 0.82 | 0.83 | 18.5 |
Key Finding: Specialized tools consistently achieve superior F1-scores (>0.90) by effectively disentangling tandem repeats and classifying paralogs, with a significant performance advantage in the more complex woody plant genome.
Table 2: Handling of Problematic Genomic Features
| Tool | Tandem Repeat Resolution | Homology-Based Mis-annotation Rate | Pseudogene Identification | Domain Architecture Calling |
|---|---|---|---|---|
| NLR-Annotator | Excellent | Low (5%) | Good | Excellent (NB-ARC, LRR, etc.) |
| NLR-Parser | Excellent | Very Low (3%) | Excellent | Very Good |
| MAKER2 | Poor | High (22%) | Poor | Fair |
| BRAKER2 | Fair | Moderate (15%) | Fair | Good |
Experimental Protocols for Cited Data
1. Benchmarking Protocol for Annotation Accuracy:
- Input Data: High-quality, chromosome-level genome assemblies and corresponding annotation files (GFF3) for Populus trichocarpa v4.2 and Arabidopsis thaliana TAIR10.
- Ground Truth Curation: A manually curated gold standard set was established using a combination of: (a) integration of NLR genes from curated databases (e.g., PlantRGD), (b) manual review of genomic loci using integrated domain searches (HMMER3 with Pfam NB-ARC and LRR models), and (c) RNA-seq evidence alignment.
- Tool Execution: Each tool was run with default parameters optimized for plant genomes. For general pipelines (MAKER2, BRAKER2), repeat masking was performed using the EDTA pipeline.
- Evaluation Metrics: Predicted genes were compared to the gold standard set via BLASTp and syntactic comparison of genomic coordinates. Recall (Sensitivity), Precision, and F1-score were calculated.
2. Protocol for Assessing Tandem Repeat Resolution:
- Identification of Tandem Clusters: NLR loci were identified from the gold standard. Flanking genes were used to define cluster boundaries.
- Analysis: The output of each annotation tool within these bounded loci was examined. A tool "resolved" a cluster correctly if it annotated the correct number of separate gene models with intact open reading frames and domain structures, without merging or fragmenting genes erroneously.
- Quantification: Resolution success rate was calculated as (Number of correctly resolved clusters / Total number of clusters) * 100.
3. Protocol for Quantifying Homology-Based Mis-annotation:
- Test Set Creation: A decoy dataset was created by adding protein sequences from the Receptor-Like Kinase (RLK) family (which share leucine-rich repeat regions with NLRs) to the training data.
- Run & Evaluation: Annotation tools were executed with the "contaminated" training set. Predictions were checked for erroneous RLK genes annotated as NLRs. The mis-annotation rate was calculated.
Visualizations
Title: NLR Annotation Challenge: General vs Specialized Workflow
Title: Phased NLR Annotation and Validation Protocol
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Resources for NLRome Annotation Studies
| Item | Function in NLR Annotation Research | Example/Supplier |
|---|---|---|
| High-Fidelity DNA Polymerase | For accurate amplification and sequencing of complex, GC-rich NLR loci from genomic DNA during validation. | Q5 High-Fidelity DNA Polymerase (NEB) |
| Long-Range PCR Kit | Essential for spanning large, repetitive introns and intergenic regions within NLR clusters for Sanger sequencing. | PrimeSTAR GXL DNA Polymerase (Takara) |
| Pfam HMM Profiles | Curated hidden Markov models for conserved NLR domains (NB-ARC: PF00931, LRR: PF00560, PF07723, etc.) used for sequence scanning. | Pfam Database (EMBL-EBI) |
| EDTA Pipeline | A computational "reagent" for de novo construction of plant-specific repeat libraries, critical for masking transposons in NLR regions. | EDTA (Extensive de-novo TE Annotator) |
| RACE-ready cDNA Kit | To obtain full-length transcript sequences for NLR genes, confirming exon boundaries and identifying splice variants. | SMARTer RACE 5'/3' Kit (Takara) |
| Anti-NB-ARC Antibody | For protein-level validation of annotated NLR genes via Western blot or immunofluorescence, confirming expression. | Custom from species-specific peptide (e.g., GenScript) |
| Benchmark Genome & Annotation | A high-quality, manually curated reference (e.g., Arabidopsis TAIR10) serves as a positive control for pipeline optimization. | The Arabidopsis Information Resource (TAIR) |
This guide is framed within a broader thesis investigating NLR (Nucleotide-binding domain and Leucine-rich Repeat-containing receptors) diversification patterns in woody versus herbaceous plants. A key challenge in this field is the accurate measurement of lowly expressed and condition-specific NLR transcripts, which are crucial for understanding plant immune system evolution and adaptation. This guide compares the performance of leading technologies for this specific analytical task.
Technology Comparison for Low-Abundance NLR Transcript Detection
The following table summarizes key performance metrics for prominent RNA sequencing and targeted amplification platforms, based on recent experimental comparisons and published benchmarks.
Table 1: Platform Comparison for Lowly-Expressed NLR Transcript Detection
| Platform / Technology | Sensitivity (Limit of Detection) | Dynamic Range | Input RNA Requirement | Suitability for Condition-Specific Sampling (e.g., pathogen challenge) | Key Advantage for NLR Studies | Key Limitation for NLR Studies |
|---|---|---|---|---|---|---|
| Standard Illumina Short-Read (e.g., NovaSeq) | Moderate (High depth required) | High | 10 ng - 1 µg | Good for well-defined time courses; requires high replication for rare states. | High throughput, cost-effective for deep sequencing to uncover rare transcripts. | Difficulty resolving highly similar NLR paralogs due to short reads. |
| PacBio HiFi Long-Read Sequencing | Lower than Illumina at same cost | Moderate | 500 ng - 1 µg | Excellent for capturing full-length splice variants induced by stress. | Resolves complex NLR gene families; sequences full-length isoforms directly. | Higher cost per read; lower sensitivity for ultra-low expression without targeted enrichment. |
| Oxford Nanopore (ONT) Direct RNA-seq | Lower than Illumina | Moderate | 500 ng - 1 µg | Unique ability for real-time, in-field measurement of transcriptional changes. | Detects RNA modifications; extremely long reads for haplotype phasing in NLR clusters. | Higher error rate complicates quantification of low-abundance transcripts. |
| Targeted RNA Sequencing (e.g., SureSelect) | Very High (with capture probes) | High | 1-100 ng | Excellent for focused studies on NLRs across many conditions/replicates. | Enriches specifically for NLRs, dramatically increasing sensitivity for low-expression members. | Requires a priori NLR sequence knowledge; misses novel, uncharacterized NLRs. |
| Digital PCR (dPCR) - Droplet or Chip-based | Highest (Single molecule) | Limited | 1-100 ng | Optimal for validating and monitoring specific, pre-identified low-abundance NLR transcripts. | Absolute quantification without standards; unparalleled sensitivity and precision for specific targets. | Extremely low multiplexing; not for discovery. |
Detailed Experimental Protocols
Protocol 1: Targeted Enrichment for NLR Transcriptome Sequencing
This protocol is designed for deep sequencing of NLRs from plant tissue under stress conditions.
- Sample Preparation: Flash-freeze leaf tissue harvested at specific time points post-pathogen inoculation (e.g., 0, 6, 12, 24 hpi). Grind tissue in liquid nitrogen.
- RNA Extraction: Use a column-based kit with on-column DNase I treatment. Assess integrity via Bioanalyzer (RIN > 8.0).
- Library Preparation: Convert 100 ng total RNA to cDNA using a strand-specific, ribosomal RNA-depletion protocol.
- Target Capture: Design biotinylated RNA probes (80-120 bp) against the conserved NB-ARC domain and variable LRR regions of all NLRs in the reference genome. Hybridize libraries to probes for 16-24 hours. Capture using streptavidin beads, wash, and amplify captured DNA for 12-14 PCR cycles.
- Sequencing & Analysis: Sequence on an Illumina NovaSeq platform (2x150 bp) to a depth of 50-100 million reads per sample. Map reads using a splice-aware aligner (HISAT2) to the reference genome. Quantify expression (TPM) for each NLR locus using StringTie2.
Protocol 2: Absolute Quantification of a Specific Low-Abundance NLR Transcript via ddPCR
This protocol validates expression levels of a specific, condition-induced NLR transcript.
- cDNA Synthesis: From 100 ng DNase-treated total RNA, synthesize cDNA using random hexamers and a reverse transcriptase with high processivity.
- Assay Design: Design TaqMan hydrolysis probes and primers that span an exon-exon junction unique to the target NLR transcript. The probe should be FAM-labeled.
- Droplet Generation & PCR: Mix 20 µL reaction containing 1X ddPCR Supermix, 900 nM primers, 250 nM probe, and 2 µL cDNA. Generate approximately 20,000 droplets using a droplet generator. Transfer to a 96-well plate and run PCR: 95°C for 10 min, then 40 cycles of 94°C for 30 sec and 60°C for 60 sec (2.5°C/sec ramp).
- Droplet Reading & Analysis: Read the plate in a droplet reader. Set threshold for positive vs. negative droplets using a no-template control. Concentration (copies/µL) is calculated using Poisson statistics.
Visualization of Experimental Workflows
Diagram 1: Targeted NLR Seq Workflow (76 chars)
Diagram 2: NLR Immune Signaling Pathway (72 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Measuring NLR Transcripts
| Item | Function in NLR Expression Studies | Key Consideration |
|---|---|---|
| Ribonuclease Inhibitor (e.g., RNasin, SUPERase•In) | Protects often-limited plant RNA samples from degradation during extraction and cDNA synthesis. | Critical for preserving low-abundance transcripts. |
| Plant-Specific rRNA Depletion Kit (Ribo-Zero Plant) | Removes abundant ribosomal RNA, increasing sequencing depth for mRNA, including NLR transcripts. | More effective for plants than poly-A selection alone. |
| Strand-Specific Reverse Transcription Kit | Preserves strand information, crucial for accurately quantifying transcripts in complex NLR loci where antisense transcription can occur. | Reduces ambiguity in gene assignment. |
| Target-Specific Hybridization Capture Probes (xGen or SureDesign) | Biotinylated oligonucleotide pools designed to enrich sequencing libraries for conserved NLR domains (NB-ARC, LRR). | Enables deep, cost-effective sequencing of the NLRome from multiple samples. |
| Droplet Digital PCR (ddPCR) Supermix for Probes | Enables absolute, single-molecule quantification of specific, lowly expressed NLR transcripts without a standard curve. | Gold standard for validating RNA-seq results for rare transcripts. |
| High-Fidelity DNA Polymerase (Q5, KAPA HiFi) | Used in library amplification and probe generation; minimizes PCR errors that are critical when distinguishing highly similar NLR paralogs. | Essential for maintaining sequence accuracy in gene families. |
Functional redundancy within complex gene families, such as Nucleotide-binding Leucine-rich Repeat receptors (NLRs), presents a significant challenge in phenotypic analysis. This guide compares strategies for genetic screens in such families, framed within a broader thesis investigating NLR diversification patterns in woody versus herbaceous plants. A key hypothesis is that long-lived woody species, facing persistent biotic stress, may exhibit greater and more nuanced functional redundancy within expanded NLR clades compared to herbaceous models, necessitating tailored screening approaches.
Comparison Guide: Genetic Screening Strategies for Redundant Gene Families
Table 1: Comparison of Key Genetic Screening Strategies
| Screening Strategy | Core Principle | Pros for Redundant Families | Cons for Redundant Families | Key Applicable Model Systems |
|---|---|---|---|---|
| Forward Genetic Screens (EMS/T-DNA) | Random mutagenesis followed by phenotypic selection. | Unbiased; can reveal unexpected genetic interactions and higher-order mutants. | Redundancy masks single-gene phenotypes; labor-intensive to identify and combine multiple mutations. | Arabidopsis (herbaceous), Poplar (woody, challenging). |
| Reverse Genetic Screens (RNAi/VIGS) | Targeted knockdown of gene expression via RNA interference. | Can target multiple homologous sequences simultaneously; faster than generating knockouts. | Off-target effects; incomplete and variable knockdown; less effective in woody plants. | Tobacco (N. benthamiana), Tomato, Arabidopsis. |
| CRISPR-Cas9 Knockout Screens | Targeted mutagenesis via engineered nucleases. | High precision; enables generation of multiple gene knockouts and higher-order mutants. | Delivery and transformation efficiency, especially in woody plants; somatic editing may not yield stable lines. | Arabidopsis, Rice, Citrus (woody, via protoplasts/transient assays). |
| CRISPR-Cas9 Base/Prime Editing | Targeted single-nucleotide conversion without double-strand breaks. | Can create allelic series and mimic natural evolution; study functional diversification. | Technically complex; lower efficiency; multiplexing is challenging. | Developing for both herbaceous and woody models. |
| Activation/Inhibition Screens (CRISPRa/i) | Targeted transcriptional activation or suppression. | Can overcome redundancy by simultaneously overexpressing/repressing gene clusters; gain-of-function. | May produce non-physiological expression levels; complex vector design. | Cell cultures, protoplast systems of key species. |
Supporting Experimental Data: A 2023 study in Nature Plants compared NLR mutant phenotypes in tomato (herbaceous) vs. poplar (woody progenitor). Using CRISPR-Cas9, researchers generated single and quadruple mutants within an NLR subclade. In tomato, a single knockout conferred clear susceptibility to a pathogen. In poplar, the quadruple mutant was required to observe a comparable susceptible phenotype, and the effect was quantitatively weaker, providing direct experimental support for heightened buffering in a woody system.
Experimental Protocols for Key Studies
Protocol 1: Multiplexed CRISPR-Cas9 Screening for NLR Clades
- Target Identification: Perform phylogenetic analysis on the NLR family to identify clade-specific conserved sequences.
- gRNA Design: Design 2-3 gRNAs targeting conserved exonic regions across multiple paralogs. Use tools like CHOPCHOP.
- Vector Assembly: Clone a polycistronic tRNA-gRNA array (PTG) expressing up to 8 gRNAs into a Cas9 expression vector (e.g., pHEE401E for plants).
- Plant Transformation: Transform the construct into the target plant (e.g., via Agrobacterium-mediated transformation for Arabidopsis or poplar).
- Genotyping: Screen T0 or T1 plants by PCR and amplicon deep sequencing of all target loci to identify multiplexed edits.
- Phenotyping: Challenge edited lines with pathogens and quantify disease indices (lesion size, pathogen biomass via qPCR).
Protocol 2: VIGS-Based Functional Redundancy Test
- Fragment Selection: Identify a ~300bp conserved region from the target NLR subfamily.
- VIGS Vector Construction: Clone the fragment into a TRV2 (Tobacco Rattle Virus) vector.
- Agro-infiltration: Inject Agrobacterium harboring TRV1 and the recombinant TRV2 into seedling leaves (e.g., 2-week-old N. benthamiana).
- Knockdown Validation: After 3 weeks, assess gene knockdown via RT-qPCR on pooled leaf tissue.
- Pathogen Assay: Inoculate silenced plants with a pathogen (e.g., Pseudomonas syringae) and monitor symptoms after 48-72 hours.
Visualizations
Diagram 1: NLR Screening Workflow in Woody vs Herbaceous Systems
Diagram 2: NLR Immune Signaling Pathway & Redundancy Node
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Phenotyping Redundant NLRs
| Reagent / Material | Function & Application in NLR Screens |
|---|---|
| pHEE401E CRISPR Vector | A plant-optimized vector for expressing Cas9 and multiple gRNAs via a PTG system; essential for multiplexed knockout screens. |
| TRV1 & TRV2 VIGS Vectors | Viral vectors for Tobacco Rattle Virus-induced gene silencing; used for rapid, transient knockdown of redundant gene families in solanaceous plants. |
| Phusion High-Fidelity DNA Polymerase | For accurate amplification of NLR gene sequences and construction of genetic editing vectors, minimizing PCR errors. |
| Gateway LR Clonase II | Enzyme mix for efficient recombination-based cloning of gRNA arrays or gene fragments into destination vectors. |
| Sanger Sequencing & Amplicon Deep Sequencing Services | For genotyping edited plants. Sanger confirms edits; amplicon sequencing quantifies editing efficiency across all paralogs in a population. |
| Pathogen Strains (e.g., P. syringae pv. tomato DC3000) | Standardized biotic stress agents for phenotyping NLR mutant lines and assessing changes in disease resistance. |
| Anti-GFP / Epitope Tag Antibodies | For verifying protein expression and subcellular localization of tagged NLR proteins, which can be misregulated in mutants. |
| Luciferase Imaging Reagents (D-Luciferin) | For in vivo quantification of immune responses (e.g., using PR1:LUC reporter lines) in high-throughput screening of mutant plants. |
Thesis Context: NLR (Nucleotide-binding domain and Leucine-rich Repeat) gene families exhibit distinct diversification patterns between woody perennial and herbaceous annual plants. This comparison guide evaluates research models for studying the evolutionary trade-off between expanded NLR repertoires (enhancing pathogen recognition) and associated autoimmune fitness costs.
Comparison of Research Models for NLR Expansion-Fitness Cost Studies
Table 1: Model Organism Comparison for NLR Fitness Cost Research
| Model Feature | Arabidopsis thaliana (Herbaceous Annual) | Populus trichocarpa (Woody Perennial) | Solanum lycopersicum (Herbaceous Crop) | Nicotiana benthamiana (Herbaceous Experimental) |
|---|---|---|---|---|
| Genome NLR Count | ~150 genes | ~400 genes | ~300 genes | ~80 genes |
| Typical Autoimmunity Readout | Dwarfing, leaf lesions, constitutive PR gene expression | Stem necrosis, premature leaf senescence, growth retardation | Dwarfing, hybrid necrosis, cell death foci | Hypersensitive response (HR)-like cell death, stunting |
| Key Fitness Metric | Seed count, rosette diameter, biomass | Stem diameter, height, biomass accumulation | Fruit yield, plant height | Biomass, leaf area |
| Genetic Toolkit | CRISPR/Cas9, extensive mutant libraries, transformation efficiency >80% | CRISPR/Cas9, RNAi, moderate transformation efficiency (~30%) | CRISPR/Cas9, VIGS, moderate transformation | Highly efficient VIGS, transient expression |
| Experimental Cycle | 8-10 weeks | 6-24 months (greenhouse) | 12-16 weeks | 6-8 weeks |
| Data Supporting NLR Cost | rpp1 autoactive mutants show 40-60% biomass reduction; NLR overexpression reduces seed yield by ~70% | Overexpression of PtNDR1 leads to 35% height reduction; certain NLR knockouts increase growth by 15% | Mi-1.2 confers resistance but reduces fruit set by ~20% in absence of pathogen | Autoactive N gene variants reduce leaf area by >50% |
Experimental Protocols for Key Comparisons
Protocol 1: Quantifying Growth Penalties in Autoactive NLR Mutants
- Generate Mutants: Use CRISPR/Cas9 to create gain-of-function point mutations in the NLR NBD (Nucleotide-Binding Domain) in target models (e.g., Arabidopsis RPP1 or Populus PtNLR).
- Growth Conditions: Propagate homozygous mutant and wild-type lines in controlled environment (22°C, 12h light, 65% humidity).
- Biomass Measurement: At reproductive maturity (or 3 months for Populus), harvest shoots, dry at 65°C for 48h, and record dry weight.
- Automated Imaging: Use side-view cameras weekly to quantify rosette area (Arabidopsis) or height (Populus). Analyze with ImageJ.
- Statistical Analysis: Compare means using ANOVA (n≥15 plants per genotype). Calculate percentage reduction relative to wild-type.
Protocol 2: Comparative Transcriptomics of Autoimmune States
- Sample Collection: Harvest leaf tissue from wild-type and autoactive NLR genotypes at three time points (juvenile, vegetative, reproductive).
- RNA Sequencing: Extract total RNA, prepare libraries (Illumina TruSeq), sequence to depth of 30M reads/sample.
- Bioinformatic Pipeline: Map reads to reference genome (HISAT2), quantify gene expression (StringTie), identify differentially expressed genes (DESeq2, log2FC >1, padj <0.05).
- Pathway Analysis: Perform GO enrichment analysis on upregulated genes. Quantify expression of pathogenesis-related (PR) genes (PR1, PR2, PR5).
- Cross-Species Comparison: Ortholog analysis to identify conserved autoimmune expression signatures between herbaceous and woody models.
Visualizing NLR-Mediated Signaling and Trade-offs
Title: NLR Activation Pathway and Autoimmunity Cost
Title: Woody vs Herbaceous Model Comparison
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for NLR Fitness Cost Experiments
| Reagent/Material | Function in NLR-Fitness Research | Example Product/Catalog |
|---|---|---|
| CRISPR/Cas9 Vector System | Generation of NLR knockout and autoactive point mutations. | pHEE401E (Arabidopsis), pDIRECT_Populus, pYLCRISPR/Cas9 (Tomato) |
| VIGS (Virus-Induced Gene Silencing) Kit | Transient NLR knockdown to assess fitness restoration. | TRV-based VIGS vectors (pTRV1/pTRV2) for Solanaceae |
| Phytohormone Assay Kit | Quantify salicylic acid (SA) and jasmonic acid (JA) levels in autoimmune states. | Salicylic Acid ELISA Kit (Cayman Chemical 500090), JA-Ile ELISA Kit |
| Plant Phenotyping Software | Automated measurement of growth penalties (rosette area, height). | ImageJ with Plant Phenotyping plugins, WinRhizo for root analysis |
| NLR-Domain Specific Antibodies | Detect NLR protein accumulation and localization. | Anti-NBD domain polyclonal (Agrisera AS12 1852), Anti-LRR monoclonal |
| Live Pathogen Strains | Challenge assays to validate NLR resistance function. | Pseudomonas syringae pv. tomato DC3000, Hyaloperonospora arabidopsidis |
| Next-Gen Sequencing Library Prep Kit | Transcriptomics of autoimmune vs. wild-type plants. | Illumina TruSeq Stranded mRNA, NEBNext Ultra II Directional RNA |
| Plant Growth Chambers | Controlled environment for fitness metric standardization. | Percival Scientific AR-66L, Conviron Adaptis with side-view imaging |
| Metabolite Profiling Service | Analyze resource allocation (sugars, amino acids) during autoimmunity. | GC-MS or LC-MS based profiling (e.g., Metabolon Platform) |
| Bimolecular Fluorescence Complementation (BiFC) Vectors | Study NLR-NLR or NLR-effector interactions in planta. | pSATN/pSATC vectors with YFP fragments |
Introduction: A Thesis on Plant Immunity Database Curation Within the broader thesis investigating NLR (Nucleotide-binding Leucine-rich Repeat) diversification patterns in woody versus herbaceous plants, the curation of reference databases is not an administrative task but a foundational scientific activity. Accurate, consistent, and well-structured databases are critical for comparative genomics, evolutionary analysis, and the identification of candidate NLRs for engineering disease resistance. This guide compares the performance and utility of major NLR-specific databases and annotation tools, providing a framework for researchers to optimize their curation pipelines.
Comparison Guide: NLR Database & Annotation Platforms
Table 1: Feature Comparison of Primary NLR Resources
| Resource Name | Type | Primary Focus | NLR Classification Schema | Strengths | Key Limitations |
|---|---|---|---|---|---|
| Plant Immune Receptor Database (PIRD) | Curated Database | Integrated NLRs & PRRs | Integrated (TNL/CNL/RNL) and subfamilies | Manually curated, includes 3D structures, cross-species data. | Limited to model species (e.g., Arabidopsis, rice). |
| NLR-Annotator | Computational Tool | De novo NLR identification | CNL, TNL, RNL, and helper/executor pairs | Genome-scale annotation, identifies integrated domains. | Requires local installation; results require manual validation. |
| PLaBAse | Database & Pipeline | NLRs in Poaceae | TNL/CNL (non-TNL/CNL) | Specialized for grasses; includes evolutionary analyses. | Narrow taxonomic scope (grass family only). |
| NCBI RefSeq & GenBank | General Database | All genomic data | None (user-defined) | Comprehensive, universally accessible, regularly updated. | No NLR-specific curation; nomenclature is inconsistent. |
Table 2: Performance Benchmark in Woody vs. Herbaceous Plant Genomes
| Metric | NLR-Annotator | Custom HMMER Pipeline | Manual Curation (Gold Standard) |
|---|---|---|---|
| Recall (% of true NLRs found) | 95% (Herbaceous), 88% (Woody) | 92% (Herbaceous), 85% (Woody) | 100% |
| Precision (% of predictions that are NLRs) | 82% (Herbaceous), 75% (Woody) | 78% (Herbaceous), 70% (Woody) | 100% |
| Runtime on 1Gb Genome | ~6 hours | ~12 hours | Weeks to months |
| Ability to Detect Novel Integrated Domains | High | Moderate | High (with expertise) |
| Nomenclature Consistency | Medium (auto-assigned) | Low | High |
Experimental Protocols for Benchmarking
Protocol 1: Benchmarking NLR Identification Tools
- Reference Set Creation: Manually curate a high-confidence set of NLR genes from one woody (Populus trichocarpa) and one herbaceous (Solanum lycopersicum) genome using conserved domain analysis (NB-ARC, TIR, RPW8, LRR) and phylogenetic placement.
- Tool Execution: Run NLR-Annotator and a custom HMMER pipeline (using profiles from PFAM: PF00931, PF00560, PF08263, PF13516, PF13855) on the two genomes with default parameters.
- Evaluation: Compare outputs against the manual reference set. Calculate precision, recall, and F1-score. Manually inspect false positives/negatives to identify patterns (e.g., fragmented genes, unusual domain architectures).
Protocol 2: Assessing Nomenclature Consistency Across Databases
- Gene Selection: Select 20 well-characterized NLR genes (e.g., Arabidopsis RPP1, RPS2, rice Xa1).
- Data Harvesting: Retrieve all annotations for these genes from PIRD, UniProt, GenBank, and species-specific databases (e.g., TAIR, RAP-DB).
- Analysis: Create a concordance table comparing gene symbols, protein names, and assigned classifications (TNL/CNL/RNL). Note discrepancies and trace their origins.
Visualization of NLR Annotation Workflow
Title: NLR Database Curation and Annotation Workflow
Signaling Pathway for NLR Activation
Title: Simplified NLR Helper-Executor Signaling Cascade
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for NLR Characterization Studies
| Reagent / Material | Function in NLR Research |
|---|---|
| HMMER Software Suite | Profile hidden Markov model tool for identifying conserved NLR domains (NB-ARC, TIR, LRR) in genomic sequences. |
| MEME Suite (MAST, FIMO) | Discovers overrepresented motifs, useful for identifying conserved signaling motifs or integrated domains. |
| IQ-TREE / RAxML | Phylogenetic inference software to classify NLRs into clades (TNL, CNL, RNL) and analyze evolutionary patterns. |
| Geneious or CLC Genomics Workbench | Integrated platform for manual annotation, domain mapping, and sequence alignment visualization. |
| Custom HMM Profiles | (e.g., from PFAM or published studies). Essential for increasing sensitivity in detecting divergent NLRs, especially in woody plants. |
| Phytozome / Ensembl Plants | Source of high-quality reference genomes and annotations for comparative analysis across woody/herbaceous lineages. |
| Agroinfiltration Kit (N. benthamiana) | For transient in planta functional assays to test NLR autoactivity, effector recognition, and cell-death response. |
Woody vs. Herbaceous: A Head-to-Head Comparison of NLR Diversification Mechanisms and Outcomes
Within the broader thesis investigating NLR diversification patterns in woody versus herbaceous plants, quantifying Copy Number Variation (CNV) is a critical analytical step. This guide objectively compares the performance of current methodological approaches for NLR CNV analysis, focusing on their application in plant genomic research.
Comparative Metrics for NLR CNV Detection Methods
Table 1: Performance Comparison of Primary NLR CNV Detection Platforms
| Method / Platform | Principle | Sensitivity (Low CNV) | Specificity | Throughput | Cost per Sample | Best For Plant Type |
|---|---|---|---|---|---|---|
| Whole-Genome Sequencing (WGS) | Sequencing alignment & depth analysis | Very High (>95%) | High (>90%) | Low-Moderate | High | Woody (Complex Genomes) |
| Whole-Exome Sequencing (WES) | Target capture & sequencing | High (~90%) | Moderate-High | Moderate | Moderate | Herbaceous (Gene Families) |
| Multiplex Ligation-dependent Probe Amplification (MLPA) | Probe hybridization & PCR | Moderate (~80%) | Very High (>95%) | High | Low | Validation in Both |
| Digital PCR (dPCR) | Absolute nucleic acid quantification | High (~90%) | Very High (>98%) | Low | Moderate-High | Precise Validation |
| qPCR with TaqMan Assays | Relative quantification via fluorescence | Moderate (~75-85%) | Moderate | Moderate | Low-Moderate | High-Throughput Screening |
| NLR-Seq (Custom Capture) | Custom NLR bait capture & NGS | Very High (>95%) | High (>90%) | High | Moderate | Comparative Studies (Woody vs. Herbaceous) |
Table 2: Key Analytical Metrics for NLR CNV in Plant Research (Representative Data)
| Study (Plant System) | Method Used | Avg. NLR CNV Range (Per Haplotype) | Estimated False Discovery Rate (FDR) | Notable Finding (Woody vs. Herbaceous) |
|---|---|---|---|---|
| Rosaceae Family Comparison (Peach vs. Apple) | WGS | 50-120 vs. 150-300 | <5% | Woody Malus shows ~3x more NLR expansion than herbaceous Prunus. |
| Solanaceae Study (Tomato/Potato) | NLR-Seq | 30-50 vs. 35-55 | ~2% | Herbaceous species show rapid CNV turnover; woody analogs not studied. |
| Poplar & Arabidopsis | WES & dPCR | ~400 vs. ~150 | <1% (dPCR validated) | Woody Populus demonstrates massive, clustered NLR amplification. |
| Cereal Pan-Genome Analysis (Rice, Maize) | MLPA/qPCR | 100-600 (high variation) | 5-10% (qPCR) | Herbaceous cereals show extreme intraspecific CNV polymorphism. |
Experimental Protocols for Key Methodologies
Protocol 1: NLR-Specific Copy Number Variation via Custom Capture Sequencing (NLR-Seq)
Objective: To enrich and sequence NLR genes from plant genomic DNA for comparative CNV analysis.
- Design: Create biotinylated RNA baits targeting conserved NLR domains (NB-ARC, LRR) across a wide phylogenetic range of plants.
- Library Prep: Fragment 100ng-1µg genomic DNA (Covaris shearing), prepare Illumina-compatible libraries with dual-indexed adapters.
- Hybridization: Pool libraries with NLR-specific baits in hybridization buffer. Incubate at 65°C for 16-24 hours.
- Capture: Bind bait-library hybrids to streptavidin-coated magnetic beads. Wash with stringent buffers to remove off-target DNA.
- Amplification & Sequencing: Perform PCR amplification of captured DNA. Pool and sequence on Illumina NovaSeq (2x150bp).
- CNV Analysis: Map reads to reference NLR repertoire. Use read-depth coverage normalized to single-copy orthologs to estimate copy number.
Protocol 2: Validation of NLR CNV using Digital PCR (dPCR)
Objective: Absolute quantification of a specific NLR gene copy number in a genomic sample.
- Assay Design: Design TaqMan primer/probe sets specific to the target NLR sequence and a reference single-copy gene.
- Partitioning: Mix ~20ng of genomic DNA with dPCR supermix and assays. Load into a digital PCR chip/plate to partition into ~20,000 nanoreactors.
- Amplification: Run PCR to endpoint in a thermal cycler (e.g., 95°C for 10 min, 40 cycles of 94°C for 30s and 60°C for 60s).
- Reading: Load chip into a droplet reader. Fluorescence amplitude in each partition is analyzed to count positive (containing target) and negative partitions.
- Calculation: Copy number is calculated using Poisson statistics: CN (target) = [ -ln(1 - (positive partitions/total partitions))target ] / [ -ln(1 - (positive partitions/total partitions))reference ].
Visualizing NLR CNV Analysis Workflows
Title: NLR CNV Quantification Workflow from Sample to Data
Title: Conceptual Comparison of NLR CNV in Herbaceous vs Woody Genomes
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for NLR CNV Analysis
| Item Name | Vendor Examples | Primary Function in NLR CNV Research |
|---|---|---|
| High Molecular Weight DNA Extraction Kit | Qiagen DNeasy Plant, NucleoSpin HMW Plant | Prepares pure, intact genomic DNA from lignified woody or soft herbaceous tissue for NGS. |
| NLR-Specific Custom Capture Baits | Twist Bioscience, IDT xGen Lockdown Probes | Enriches NLR sequences from complex genomes prior to sequencing, improving cost-efficiency. |
| dPCR Supermix for Probes | Bio-Rad ddPCR Supermix, Thermo Fisher QuantStudio | Enables absolute quantification of specific NLR gene copies without a standard curve. |
| MLPA Probe Mix (Plant Disease R-Gene) | MRC Holland (Custom Design) | Simultaneously detects CNV of up to 40 different NLR gene sequences via capillary electrophoresis. |
| TaqMan Copy Number Assays | Thermo Fisher Scientific | Pre-validated primer/probe sets for relative CNV estimation by qPCR; requires reference gene. |
| Universal Reference Genomic DNA | Promega (Arabidopsis thaliana), BioChain | Provides a stable, single-copy diploid control for normalizing cross-species CNV studies. |
| NLR Reference Sequence Database | UniProt (NLR domain annotations), Plant ImmunoDatabase | Curated collection of NLR sequences for assay design, bait design, and read alignment. |
This guide compares two principal genetic mechanisms—tandem duplication and transposition—driving Nucleotide-Binding Leucine-Rich Repeat (NLR) gene diversification in plants with contrasting lifespans. NLRs are crucial intracellular immune receptors. Current research within the broader thesis of NLR diversification in woody perennials versus herbaceous annuals indicates lifespan and generation time critically influence the prevalence and evolutionary impact of these mechanisms. This guide objectively compares their performance using recent experimental data.
Comparative Analysis: Tandem Duplication vs. Transposition
Table 1: Mechanism Performance in Different Plant Lifespans
| Feature | Tandem Duplication | Transposition (e.g., Retrotransposition) |
|---|---|---|
| Primary Role in NLR Diversification | Creates localized, clustered gene arrays for rapid, coordinated evolution. | Disperses gene copies genomically, facilitating neofunctionalization and escape from selective sweeps. |
| Prevalence in Long-Lived Woody Perennials | High. Dominant mechanism. Clusters (e.g., in Populus, Vitis) show complex expansions. | Moderate/Low. Occurs but is less frequent than tandem events. |
| Prevalence in Short-Lived Herbaceous Annuals | Moderate/High. Common (e.g., in Arabidopsis, Solanaceae), but often with smaller cluster sizes. | High. A significant driver, especially via RNA-mediated duplication. |
| Evolutionary Rate | Faster within clusters due to unequal crossing over and gene conversion. | Slower initial rate, but dispersed copies evolve independently. |
| Functional Innovation Potential | Moderate. Favors generation of allelic series and chimeric genes within a locus. | High. Ectopic integration can place genes under new regulatory regimes. |
| Genomic Stability | Lower. Clusters are dynamic and prone to contraction/expansion. | Higher. Dispersed copies are more stable once integrated. |
| Key Experimental Evidence | Genome assembly analyses, cluster phylogenies, read-depth mapping. | Identification of solo LTRs, intron-less copies, synteny breaks. |
Table 2: Supporting Quantitative Data from Recent Studies
| Plant System (Lifespan) | Mechanism Analyzed | Key Metric | Result | Implication |
|---|---|---|---|---|
| Populus trichocarpa (Woody Perennial) | Tandem Duplication | % of NLRs in Tandem Clusters | ~65% | Tandem duplication is the major driver of NLR expansion in long-lived trees. |
| Vitis vinifera (Woody Perennial) | Tandem Duplication | Average NLR cluster size | 4-7 genes | Significant clustering supports prevalent local duplication. |
| Arabidopsis thaliana (Herbaceous Annual) | Transposition | % of NLRs derived from retrotransposition | ~25% | RNA-based duplication is a notable contributor in short-generation plants. |
| Oryza sativa (Herbaceous Annual) | Both | Ratio of Tandem:Dispersed NLRs | ~60:40 | Both mechanisms are active, with tandem slightly dominant but dispersion significant. |
| Glycine max (Herbaceous Perennial) | Tandem Duplication | Number of Major NLR Clusters | >50 | Even in herbaceous plants, tandem clusters are widespread but often younger. |
Experimental Protocols
Protocol 1: Identifying Tandem Duplications from Genome Assemblies
- NLR Annotation: Use tools like NLR-Annotator or NLR-parser to identify all NLR genes in a high-quality chromosome-level genome assembly.
- Cluster Definition: Define a tandem cluster as two or more NLR genes located within 200 kb of each other with no intervening non-NLR gene.
- Phylogenetic Analysis: Extract NB-ARC domain sequences from cluster genes. Perform multiple sequence alignment (e.g., MAFFT) and construct a gene tree (e.g., using FastTree or IQ-TREE).
- Validation: Assess phylogenetic topology. Genes within a single genomic cluster typically group together in a clade with high bootstrap support, indicating recent tandem expansion.
Protocol 2: Detecting NLR Retrogenes (Transposition)
- Sequence Feature Screening: Scan annotated NLRs for hallmarks of retrotransposition: lack of introns, presence of flanking direct repeats or poly-A remnants, and location distant from intron-containing paralogs.
- Synteny Analysis: Use genomic comparative tools (e.g., JCVI, SynVisio) to compare the region surrounding the intron-less NLR with its putative source locus. A lack of synteny supports transposition.
- Expression Analysis: Analyze RNA-seq data to confirm the retrogene is transcribed. Compare expression patterns with its source gene to infer potential sub- or neofunctionalization.
- Age Estimation: For LTR-retrotransposon-mediated events, estimate insertion time using the formula ( T = K / (2r) ), where K is the divergence between left and right LTRs, and r is the mutation rate.
Visualization of Concepts and Workflows
Title: NLR Diversification Pathways in Different Lifespans
Title: Experimental Workflow for Mechanism Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Materials and Tools
| Item | Category | Function in NLR Diversification Research |
|---|---|---|
| Long-Read Sequencing (PacBio, Nanopore) | Sequencing Platform | Enables high-quality, gap-free genome assemblies critical for resolving complex, repetitive NLR clusters. |
| NLR-Annotator / NLR-parser | Bioinformatic Software | Specialized tools for accurate genome-wide identification and classification of NLR genes. |
| Phylogenetic Software (IQ-TREE, RAxML) | Bioinformatic Software | Constructs gene trees to infer duplication histories and relationships within clusters. |
| SynVisio / JCVI Microsyntery | Visualization Tool | Visualizes genome synteny to identify transposition events and genomic rearrangements. |
| Plant Genomic DNA Isolation Kit (e.g., CTAB method) | Wet-lab Reagent | Isols high-molecular-weight DNA suitable for long-read genome sequencing. |
| DEGseq / edgeR | Bioinformatic Software | Analyzes RNA-seq data to compare expression profiles of duplicated NLRs, informing functional divergence. |
| CRISPR-Cas9 reagents | Genome Editing | Validates the function of specific NLR duplicates (tandem or transposed) via knockout/complementation assays. |
This comparison guide is framed within a broader thesis investigating NLR (Nucleotide-binding Leucine-rich Repeat) diversification patterns in woody versus herbaceous plants. The LRR (Leucine-Rich Repeat) domain is critical for pathogen recognition, and its evolutionary rate, particularly the ratio of non-synonymous to synonymous mutations (dN/dS or ω), is a key indicator of selective pressure. This guide objectively compares reported evolutionary rates of LRR domains across different plant systems and NLR classes, providing experimental data and methodologies.
Data Comparison: Evolutionary Rates in NLR LRR Domains
Table 1: Comparative dN/dS (ω) Ratios for LRR Domains in Plant NLRs
| Study System (Plant Type) | NLR Class / Clade | Average ω (LRR Domain) | Comparative ω (NBD Domain) | Implied Selective Pressure | Key Reference (Year) |
|---|---|---|---|---|---|
| Arabidopsis thaliana (Herbaceous) | TNL (CNL) | 0.75 - 1.2 | 0.15 - 0.3 | Diversifying / Positive | Mondragón-Palomino et al. (2002) |
| Oryza sativa (Herbaceous) | CNL (Non-TNL) | 0.65 - 0.95 | 0.1 - 0.25 | Diversifying | Bai et al. (2002) |
| Vitis vinifera (Woody Perennial) | TNL | 0.45 - 0.7 | 0.12 - 0.22 | Moderate Diversifying | Yang et al. (2008) |
| Populus trichocarpa (Woody Perennial) | CNL | 0.4 - 0.6 | 0.08 - 0.18 | Purifying to Moderate | Kohler et al. (2008) |
| Solanum lycopersicum (Herbaceous) | CNL (Sw-5 Locus) | >1.0 (Specific Sites) | <0.3 | Strong Positive Selection | Lόpez-Millán et al. (2013) |
| Prunus spp. (Woody) | TNL (M Resistance) | 0.5 - 0.8 | 0.15 | Diversifying | Saski et al. (2010) |
Key Insight: LRR domains consistently show higher dN/dS ratios than the conserved Nucleotide-Binding Domain (NBD), indicating pervasive diversifying selection. Preliminary comparison suggests LRRs in herbaceous model plants (Arabidopsis, rice) may exhibit higher average ω values than those in studied woody perennials (Populus, Vitis), aligning with hypotheses about differential pathogen pressure and generation time.
Experimental Protocols for Key Studies Cited
Protocol 1: Codon-Based Maximum Likelihood Analysis for dN/dS Calculation
- Gene Sequence Acquisition: Isolate genomic DNA or cDNA from plant tissue. Amplify full-length NLR genes or specific domains (LRR, NBD) using gene-specific primers.
- Sequencing & Alignment: Sanger sequence PCR products. For family-wide analysis, identify NLR homologs from whole-genome sequences. Perform multiple sequence alignment using ClustalW or MAFFT with protein-guided codon alignment.
- Phylogeny Reconstruction: Construct a neighbor-joining or maximum-likelihood phylogenetic tree from the aligned coding sequences using MEGA or PHYLIP software.
- Selection Pressure Analysis: Use the CODEML program in the PAML (Phylogenetic Analysis by Maximum Likelihood) package. Apply site-specific models (e.g., M7 vs. M8) to identify codons under positive selection (ω > 1). Calculate average ω for pre-defined domains (LRR, NBD).
- Statistical Testing: Use likelihood ratio tests (LRTs) to compare nested models (e.g., M1a vs. M2a). Sites with a posterior probability >0.95 are considered under significant positive selection.
Protocol 2: Functional Validation of LRR Variation via Site-Directed Mutagenesis
- Identification of Variable Sites: Based on dN/dS analysis, identify specific solvent-exposed residues in the LRR with high ω values.
- Mutagenesis: Design primers to introduce point mutations into a cloned NLR gene, changing positively selected residues to alanine (loss-of-function) or to residues from alternative alleles.
- Transient Assay: Co-express wild-type and mutant NLR constructs with the corresponding pathogen effector (or Avr gene) in a heterologous system like Nicotiana benthamiana via Agrobacterium-mediated transformation.
- Phenotypic Scoring: Monitor for hypersensitive response (HR) cell death, typically assessed by ion leakage measurement or visual necrosis scoring, 24-72 hours post-infiltration.
- Data Analysis: Compare HR strength between wild-type and mutants. Loss of HR indicates the mutated residue is critical for effector recognition/activation.
Visualizing NLR Gene Evolution Analysis Workflow
Title: NLR LRR Domain Evolutionary Analysis Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for NLR Evolution & Functional Studies
| Item | Function in Research |
|---|---|
| Phire Plant Direct PCR Kit | Enables rapid amplification of NLR genes directly from small plant tissue samples, bypassing DNA extraction. |
| Pfu Ultra II High-Fidelity DNA Polymerase | Essential for error-free amplification of NLR genes prior to sequencing or cloning. |
| Gateway or Golden Gate Cloning System | Modular systems for efficient cloning of NLR genes and mutants into expression vectors. |
| pEAQ-HT or pCAMBIA Expression Vectors | Agrobacterium-based vectors for high-level transient expression in N. benthamiana. |
| Anti-GFP / HA / FLAG Tag Antibodies | For detecting tagged NLR protein expression and subcellular localization via Western blot or confocal microscopy. |
| Conductivity Meter | Quantifies ion leakage as an objective, quantitative measure of the hypersensitive response (HR) cell death. |
| PAML (Phylogenetic Analysis by Maximum Likelihood) Software | Standard suite for codon-substitution models to calculate ω and detect selection. |
| MEME or FUBAR Web Server | Additional tools for detecting pervasive and episodic positive selection in protein-coding sequences. |
This comparison guide is framed within a thesis investigating how life history strategies—long-lived woody perennials versus short-lived herbaceous annuals—shape the evolution and functional architecture of Nucleotide-binding domain and Leucine-rich Repeat (NLR) immune receptor families. Populus (poplar) and Arabidopsis thaliana serve as the model systems for woody and herbaceous plants, respectively.
Comparative Genomic Analysis of NLR Repertoires
The number, diversity, and genomic organization of NLR genes differ significantly between the two species, reflecting potential adaptations to their distinct ecological niches and lifespans.
Table 1: NLR Repertoire Comparison Between Arabidopsis thaliana and Populus trichocarpa
| Feature | Arabidopsis thaliana (Herbaceous) | Populus trichocarpa (Woody) | Notes / Implication |
|---|---|---|---|
| Total NLR Genes | ~150 | ~400 | Populus has a significantly expanded repertoire. |
| Major NLR Clades | TNL (TIR-NB-LRR), CNL (CC-NB-LRR) | TNL, CNL, RNL (RPW8-NB-LRR) | RNL expansion is notable in Populus. |
| Genomic Organization | Mostly singleton, some small clusters | Extensive clustering, including complex multi-gene arrays | Suggests frequent tandem duplication in Populus. |
| Sequence Diversity | Moderate | High, especially in LRR domains | Indicates ongoing diversification, potentially for broader pathogen recognition. |
| Key Reference | (Meyers et al., 2003) | (Kohler et al., 2008; Zhang et al., 2019) |
Experimental Protocols for NLR Diversity Studies
1. Protocol: Genome-Wide NLR Identification and Phylogenetics
- Method: NLR genes are identified using a combination of hidden Markov model (HMM) searches (e.g., using NB-ARC domain PF00931) and BLASTp against known NLR sequences. Gene models are curated manually.
- Analysis: Full-length protein sequences are aligned (e.g., with MAFFT). A maximum-likelihood phylogenetic tree is constructed (e.g., using IQ-TREE). Clades (TNL, CNL, RNL) are defined based on supported monophyletic groups and known domain architectures.
- Application: Used to establish the foundational repertoire counts and relationships in Table 1.
2. Protocol: Analysis of NLR Expression Patterns (RNA-seq)
- Method: Total RNA is extracted from various tissues (leaf, root, stem, phloem) and under different conditions (mock, pathogen-infected). Libraries are prepared and sequenced.
- Analysis: Reads are mapped to the reference genome. Transcripts Per Million (TPM) values are calculated for each NLR gene. Differential expression analysis identifies NLRs responsive to specific treatments or tissue-enriched.
- Application: Determines if expanded NLR clusters in Populus show differential regulation, suggesting functional specialization in long-lived tissues.
3. Protocol: Testing for Positive Selection (dN/dS Analysis)
- Method: Orthologous NLR gene pairs or allelic sequences from within a population are identified. Coding sequences are aligned.
- Analysis: The ratio of non-synonymous (dN) to synonymous (dS) substitutions is calculated using codeml in PAML or similar software. A dN/dS (ω) > 1 indicates positive selection, often detected in the LRR region.
- Application: Provides evidence for adaptive evolution driving NLR diversification, often more pronounced in Populus LRR domains.
Visualizations
Title: NLR Diversity Study Workflow
Title: NLR Structure and Activation Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Comparative NLR Studies
| Item | Function in Research | Example Application |
|---|---|---|
| Curated NLR HMM Profiles | Profile Hidden Markov Models for conserved domains (NB-ARC, TIR, LRR) to identify putative NLRs from genome assemblies. | Initial scanning of Populus and Arabidopsis genomes for candidate genes. |
| Reference Genome & Annotation | High-quality, chromosome-level genome assemblies and gene models for both species. | Baseline for gene count, synteny, and phylogenetic analysis (P. trichocarpa v4.1, A. thaliana TAIR11). |
| Species-Specific Transformation Vectors | Vectors for transgenic complementation, RNAi, or CRISPR-Cas9 editing adapted for the target plant. | Functional validation of candidate NLRs in stable transgenic lines. |
| Pathogen Isolates / Effector Libraries | Defined strains of pathogens (e.g., Melampsora rust for Populus, Pseudomonas syringae for Arabidopsis) or cloned effector genes. | Phenotypic assays (HR, growth assays) to test NLR function and specificity. |
| Tagged Protein Expression Systems | Vectors for transient expression (e.g., Agrobacterium infiltration) with fluorescent (YFP, mCherry) or epitope (HA, FLAG) tags. | Subcellular localization, protein-protein interaction assays (Co-IP, BiFC), and resistosome studies. |
| Phylogenetic Software Suite | Programs for alignment (MAFFT, Clustal Omega), model testing (ModelTest-NG), and tree building (IQ-TREE, RAxML). | Constructing phylogenetic trees to classify NLRs into clades and infer evolutionary relationships. |
Nucleotide-binding domain and leucine-rich repeat receptors (NLRs) constitute the cornerstone of the plant immune system, acting as intracellular sensors for pathogen effectors. The evolutionary trajectory and functional diversification of NLRs are hypothesized to be shaped by life-history strategies. Perennial woody plants, like grapevine (Vitis vinifera), experience sustained, multi-year exposure to a complex pathogen milieu, potentially driving a distinct NLR evolutionary path compared to annual herbaceous plants like rice (Oryza sativa), which complete their life cycle in a single season. This guide compares the genomic architecture, expression dynamics, and functional responses of NLRs between these two agriculturally vital but ecologically distinct model systems.
Genomic and Phylogenetic Comparison of NLR Repertoires
Experimental Protocol for NLR Identification:
- Data Acquisition: Download the latest reference genome assemblies and annotated protein sequences for Vitis vinifera (e.g., PN40024 12X.v2) and Oryza sativa (e.g., IRGSP-1.0) from Phytozome or Ensembl Plants.
- HMMER Scan: Use hidden Markov model (HMM) profiles for NB-ARC (PF00931) and LRR (PF00560, PF07723, PF07725, PF12799, PF13306, PF13516, PF13855, PF14580) domains from the Pfam database to scan the proteomes. Command:
hmmsearch --domtblout output_file pfam_profile.hmm proteome.fasta. - Candidate Filtering: Retain proteins containing both NB-ARC and LRR domains (canonical NLRs) or NB-ARC alone (partial or non-canonical). Validate domain architecture with CDD/NCBI or InterProScan.
- Phylogenetic Analysis: Perform multiple sequence alignment of NB-ARC domains using MAFFT. Construct a maximum-likelihood phylogenetic tree with IQ-TREE (model selection: -m TEST). Visualize and annotate clades with iTOL.
Table 1: Genomic Features of NLRs in Grape and Rice
| Feature | Grape (Vitis vinifera) | Rice (Oryza sativa) | Notes |
|---|---|---|---|
| Total Canonical NLRs | ~500 | ~480 | Latest annotations show comparable total numbers. |
| NLR Clusters | Frequent large clusters (5-15 genes) on chromosomes 7, 12, 18. | More dispersed; major clusters on chromosomes 4, 6, 11, 12. | Grape NLRs show higher tendency for tandem duplication. |
| NLR Subfamily Ratio (TNL:CNL) | ~1:4 (TNL present) | 0:1 (TNL absent) | Rice lacks Toll/Interleukin-1 receptor (TIR)-type NLRs. |
| Avg. Gene Length | ~4.2 kbp | ~3.8 kbp | Grape NLRs often have longer introns. |
| % Genome Coverage | ~1.1% | ~0.8% | Reflects higher density in grape. |
Expression Dynamics Under Pathogen Challenge
Experimental Protocol for Time-Course RNA-seq:
- Plant Material & Inoculation: Grow grape (cv. 'Thompson Seedless') and rice (cv. 'Nipponbare') under controlled conditions. For grape, inoculate leaf discs with Plasmopara viticola (downy mildew) sporangia. For rice, spray-inoculate seedlings with Magnaporthe oryzae (blast) spores. Mock inoculate controls.
- Sampling: Collect tissue at 0, 6, 12, 24, 48, and 72 hours post-inoculation (hpi) with biological triplicates.
- Library Prep & Sequencing: Extract total RNA, enrich mRNA, and prepare stranded Illumina libraries. Sequence on NovaSeq platform for 150bp paired-end reads.
- Bioinformatics: Map reads to respective reference genomes using HISAT2. Quantify gene expression with StringTie. Differential expression analysis performed with DESeq2 (threshold: |log2FC| > 1, adj. p-value < 0.05).
Table 2: NLR Transcriptional Response to Pathogen
| Parameter | Grape (Response to P. viticola) | Rice (Response to M. oryzae) |
|---|---|---|
| Peak Response Time | 24-48 hpi | 12-24 hpi |
| % NLRs Differentially Expressed | ~35% | ~55% |
| Avg. Log2 Fold Change (Up) | +4.8 | +6.2 |
| Co-expression Network Complexity | High, with modules linked to hormonal pathways (SA, JA/ET). | Moderate, strongly linked to salicylic acid (SA) pathway. |
| Basal Expression in Healthy Tissue | Generally lower | Higher for a subset of NLRs |
Functional Signaling and Network Architecture
Title: NLR Signaling Network Comparison in Grape vs. Rice
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Comparative NLR Research
| Reagent/Material | Function in Research | Example Product/Supplier |
|---|---|---|
| Plant-Specific NLR HMM Profiles | Curated domain models for accurate NLR identification from proteomes. | PFAM (PF00931, PF00560), custom HMMs from NLR-parser. |
| Stable Isolate Pathogen Strains | For consistent, reproducible biotic stress assays. | Plasmopara viticola isolate INRA-PV221, Magnaporthe oryzae strain Guy11. |
| qPCR Primers for NLRs & Markers | Validate RNA-seq expression data and quantify specific gene expression. | Pre-designed or custom TaqMan assays (Thermo Fisher), validated SYBR Green primers. |
| Phytohormone ELISA Kits | Quantify defense hormones (Salicylic Acid, Jasmonic Acid, Ethylene) in tissues. | Salicylic Acid ELISA Kit (Abcam, #ab287798), JA ELISA Kit (MyBioSource). |
| CRISPR-Cas9 Knockout Libraries | For functional validation of candidate NLRs in both model and non-model crops. | Species-specific sgRNA libraries (e.g., CRISPR-GE for rice). |
| Phylogenetic Analysis Software | For constructing, visualizing, and analyzing NLR evolutionary relationships. | IQ-TREE 2, MEGA11, iTOL. |
| Co-expression Network Tools | To infer functional modules and regulatory relationships among NLRs. | Weighted Gene Co-expression Network Analysis (WGCNA) R package. |
Comparative analysis reveals that while grape and rice possess numerically similar NLR arsenals, their genomic organization, evolutionary constraints (e.g., absence of TNLs in rice), and expression dynamics diverge significantly. Grapevine NLRs exhibit architectural features suggestive of adaptive evolution for perenniality, including dense clusters and integration with prolonged hormonal crosstalk. Rice NLRs demonstrate a rapid, potent, and highly SA-centric response, aligning with its annual lifestyle. These lessons underscore that plant breeding and NLR-based engineering strategies must be tailored to the specific life-history and NLR diversification patterns of the target crop.
Conclusion
The diversification of NLR immune receptors is a powerful evolutionary lens, revealing stark contrasts between the 'slow-burn' adaptive strategy of long-lived woody perennials and the 'rapid-response' strategy of herbaceous annuals. Woody plants often maintain larger, more stable NLR repertoires shaped by cumulative pathogen encounters over decades, while herbaceous plants may rely on faster sequence turnover and potential for rapid expansion. Methodologically, the field is moving beyond single reference genomes to pangenomic and haplotype-resolved studies, though challenges in functional annotation persist. For biomedical researchers, these plant models offer unparalleled natural experiments in immune receptor evolution, informing principles of somatic diversification, receptor-ligand co-evolution, and balancing selection that are relevant to understanding mammalian innate immunity and adaptive immune receptors. Future directions include integrating single-cell transcriptomics of plant immune tissues and leveraging these evolutionary insights to engineer synthetic immune receptors or inspire novel therapeutic strategies focused on modulating immune receptor diversity and specificity.