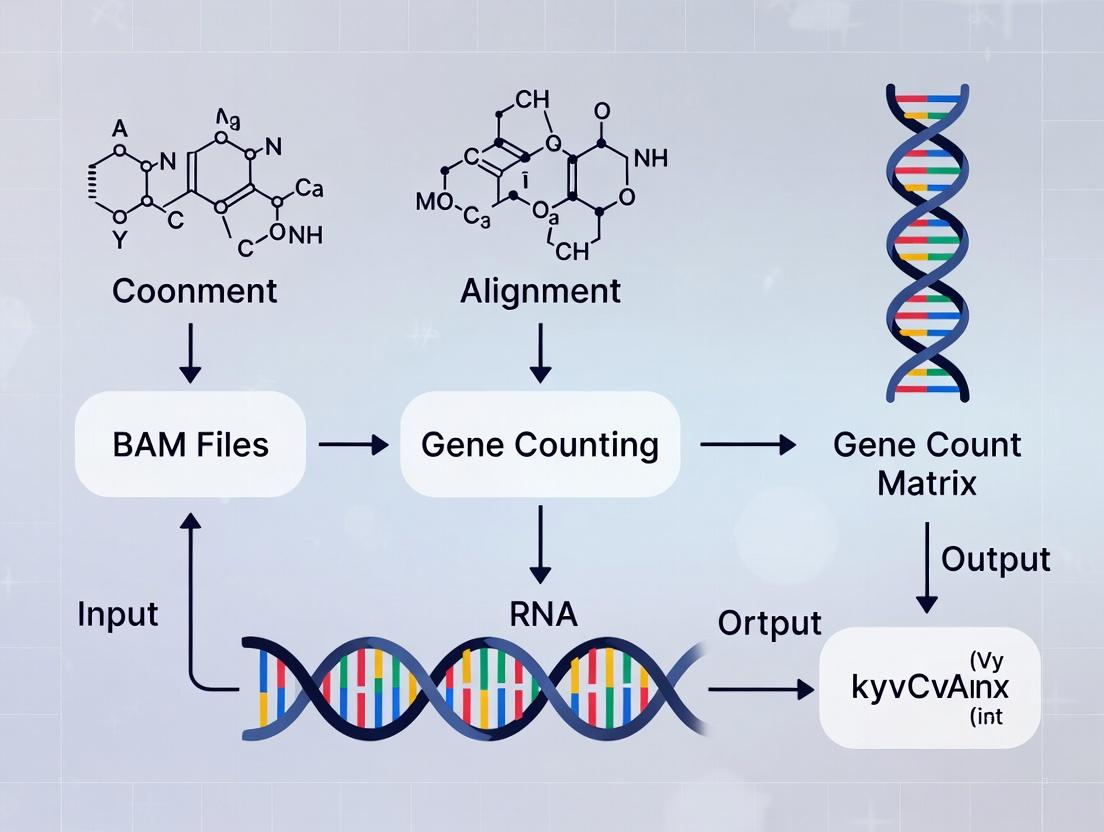
From Alignments to Insights: A Step-by-Step Guide to Creating Gene Count Matrices from BAM Files
This comprehensive guide provides researchers, scientists, and drug development professionals with a complete workflow for generating gene count matrices from BAM alignment files, a critical step in RNA-seq and other...
From Alignments to Insights: A Step-by-Step Guide to Creating Gene Count Matrices from BAM Files
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with a complete workflow for generating gene count matrices from BAM alignment files, a critical step in RNA-seq and other genomic analyses. We cover the foundational concepts of alignment files and count matrices, detail methodological workflows using leading tools like featureCounts and HTSeq, address common troubleshooting and optimization strategies, and discuss essential validation and comparative analysis techniques to ensure robust downstream results for differential expression and biomarker discovery.
Understanding the Blueprint: What Are BAM Files and Gene Count Matrices?
Within a broader thesis on generating a gene count matrix from BAM files, understanding the BAM file's genesis and structure is the foundational step. This Application Note details the critical role of the BAM file as the central, standardized output of read alignment, bridging the gap between raw sequencing data (FASTQ) and quantitative expression analysis.
From FASTQ to BAM: The Alignment Workflow
The creation of a BAM file is a multi-step computational process. The primary quantitative outputs of this pipeline are summarized in Table 1.
Table 1: Key Quantitative Metrics in Alignment & BAM Generation
| Metric | Typical Range/Value | Significance for Downstream Gene Counting |
|---|---|---|
| Overall Alignment Rate | >70-90% (species/dataset dependent) | Low rates indicate poor library quality or contamination. |
| Uniquely Mapped Reads | Ideally >80% of aligned reads | Crucial for accurate gene assignment; multi-mapped reads are ambiguous. |
| Reads Mapped in Proper Pairs | >90% for intact RNA-seq libraries | Indicates library fragment size integrity; important for transcriptome aligners. |
| Duplication Rate (PCR/optical) | Varies; >50% may be problematic | High rates reduce library complexity; marked in BAM for downstream filtering. |
| BAM File Size (vs. FASTQ) | ~0.3-0.5x of compressed FASTQ size | Binary format is highly efficient for storage and indexing. |
Protocol 1.1: Core Alignment Workflow using STAR This protocol aligns paired-end RNA-seq reads to a reference genome, producing a sorted BAM file ready for gene counting.
Prerequisite: Generate Genome Index.
STAR --runThreadN 10 --runMode genomeGenerate --genomeDir /path/to/GenomeDir --genomeFastaFiles GRCh38.primary_assembly.fa --sjdbGTFfile gencode.v44.annotation.gtf --sjdbOverhang 100--sjdbOverhang: Read length minus 1. Critical for splice junction detection.
Alignment.
STAR --runThreadN 12 --genomeDir /path/to/GenomeDir --readFilesIn sample_R1.fastq.gz sample_R2.fastq.gz --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 10 --quantMode GeneCounts --outFileNamePrefix sample_aligned.--outSAMtype BAM SortedByCoordinate: Directly outputs a coordinate-sorted BAM.--quantMode GeneCounts: STAR can output preliminary gene counts, useful for quick QC.--outFilterMultimapNmax: Limits multi-mapping reads; 10 is common default.
Post-processing (Mark Duplicates with Picard).
java -jar picard.jar MarkDuplicates I=sample_aligned.Aligned.sortedByCoord.out.bam O=sample_aligned.sorted.markdup.bam M=markdup_metrics.txt- Adds a
1024(0x400) SAM flag to duplicate reads, allowing tools like featureCounts to ignore them.
- Adds a
Diagram 1: Workflow from FASTQ to Gene Count Matrix (43 chars)
BAM File Structure: The Bridge to Gene Counting
A BAM file's internal structure directly informs the gene counting logic. The alignment section contains the critical fields used by quantification tools.
Table 2: Essential SAM/BAM Fields for Gene Counting
| SAM Field | Example | Role in Gene Count Matrix Creation |
|---|---|---|
| QNAME | NS500451:154:H5KT5AFXY:1:11101:10000:11012 |
Read identifier; used to identify read pairs. |
| FLAG | 99 (0x63) |
Binary flag indicating strand, pair info, alignment quality. |
| RNAME | chr1 |
Reference chromosome/contig name. |
| POS | 10000 |
1-based leftmost mapping position. |
| MAPQ | 255 (STAR unique) |
Mapping quality; high values (>10) favor unique mapping. |
| CIGAR | 50M100N50M |
Encodes alignment match/mismatch/insertion/deletion/splicing. |
| Tags | XS:A:+, NH:i:3, GN:Z:TP53 |
XS: Splice strand; NH: Number of alignments; GN: Gene name (some aligners). |
Protocol 2.1: Extracting and Visualizing Alignment Information
View BAM File:
samtools view -h sample.bam | head -50Check Alignment Statistics:
samtools flagstat sample_aligned.sorted.markdup.bam > sample_flagstat.txtIndex BAM File (Required by most counters):
samtools index sample_aligned.sorted.markdup.bamVisualize in IGV: Load the indexed BAM (.bam and .bai files) and the corresponding GTF annotation file into IGV to visually confirm alignments against gene models.
Diagram 2: BAM File Internal Structure (32 chars)
The Scientist's Toolkit: Essential Reagents & Software
Table 3: Key Research Reagent Solutions for BAM-Based Analysis
| Item | Function & Relevance |
|---|---|
| Reference Genome (FASTA) | The canonical sequence (e.g., GRCh38) to which reads are aligned. Foundation of the coordinate system in the BAM file. |
| Gene Annotation (GTF/GFF) | File defining genomic coordinates of genes, transcripts, and exons. The "map" used to assign aligned reads (BAM) to features. |
| Alignment Software (STAR, HISAT2) | Computational "reagent" that performs the mapping of reads to the reference, incorporating splice-aware logic for RNA-seq. |
| SAM/BAM Tools (samtools) | Essential toolkit for manipulating, sorting, indexing, and querying BAM files. |
| Duplicate Marking Tool (Picard, samtools markdup) | Identifies PCR/optical duplicates, marking them in the BAM file to prevent over-counting. |
| Gene Quantification Tool (featureCounts, HTSeq-count) | The core software that intersects BAM file coordinates with GTF annotations to produce the raw gene count matrix. |
| High-Performance Computing (HPC) Cluster/Cloud) | Alignment and BAM processing are computationally intensive, requiring significant CPU, memory, and storage. |
From BAM to Count Matrix: The Final Step
The final protocol in the context of our thesis uses the processed BAM file as direct input for quantification.
Protocol 4.1: Generating Gene Counts with featureCounts
Input: Sorted, duplicate-marked BAM file (
sample.bam) and a GTF annotation file.Run featureCounts:
featureCounts -T 8 -p --countReadPairs -a gencode.v44.annotation.gtf -o gene_counts.txt -g gene_id sample_aligned.sorted.markdup.bam-p: Count fragments (read pairs) for paired-end data.--countReadPairs: Specifically instructs to count read pairs.-g gene_id: Summarize counts at the gene level (using thegene_idattribute in GTF).
Output: The
gene_counts.txtfile contains a table where rows are genes, columns are samples, and values are the raw counts—the primary input for differential expression analysis.
Diagram 3: BAM to Count Matrix Process (33 chars)
1. Introduction: The Central Data Structure
In the broader thesis on creating a gene count matrix from BAM files, the gene count matrix is defined as the final, structured numerical table where rows represent genomic features (e.g., genes, transcripts, exons), columns represent individual biological samples (e.g., sequenced libraries), and each cell contains an integer count of sequencing reads assigned to that feature in that sample. This matrix is the indispensable input for downstream statistical analyses, such as differential expression testing with tools like DESeq2, edgeR, or limma-voom. Its accurate construction directly determines the validity of all subsequent biological conclusions.
2. Quantitative Summary of Common Counting Tools
The choice of counting tool involves trade-offs between speed, accuracy, and genomic context handling. The following table summarizes key metrics and characteristics based on recent benchmarks.
Table 1: Comparison of Feature Counting Tools (2023-2024 Benchmarks)
| Tool | Primary Method | Speed (Relative) | Accuracy Focus | Handles Ambiguous Reads | Output |
|---|---|---|---|---|---|
| featureCounts | Alignment-based | Fast | High (read-level) | Yes (via --fracOverlap) |
Count matrix |
| HTSeq | Alignment-based | Medium | High (position-based) | Yes (via overlap modes) | Count files per sample |
| Salmon / kallisto | Pseudoalignment | Very Fast | Transcript-level estimation | Model-based | Estimated counts (TPM) |
| Subread | Alignment-based | Very Fast | Junction-aware | Configurable | Count matrix |
| Qualimap | Alignment-based | Medium | Visual QC & counting | Limited | Counts & QC stats |
3. Detailed Protocol: Generating a Count Matrix from BAM using featureCounts
This protocol details the generation of a gene-level count matrix from coordinate-sorted BAM files using featureCounts (from the Subread package), a widely adopted method for its efficiency and accuracy.
Materials & Reagents:
- Input: Coordinate-sorted BAM files (one per sample), corresponding BAM index files (
.bai). - Genome Annotation: A Gene Transfer Format (GTF) file for the relevant genome assembly.
- Software: Subread package (v2.0.0 or higher) installed.
- Computing Environment: Unix/Linux command-line environment with sufficient memory (≥ 8 GB RAM for mammalian genomes).
Procedure:
- Prepare the Environment:
- Organize all BAM files in a single directory.
- Ensure the GTF file is available and corresponds to the same genome build used for read alignment.
Run featureCounts on Multiple Samples:
- Execute the following command to process all BAM files simultaneously and generate a combined matrix.
Process the Output:
- The primary output file
gene_count_matrix.txtcontains a header with meta-information, followed by the count matrix with aGeneidcolumn and counts for each BAM file. - A summary file (
gene_count_matrix.txt.summary) contains mapping statistics for each sample.
- The primary output file
Format for Downstream Analysis:
- Strip the header and first column (annotation info) to create a pure integer matrix. This can be done with command-line tools (
tail,cut) or in R/Python. - Example in R:
- Strip the header and first column (annotation info) to create a pure integer matrix. This can be done with command-line tools (
4. Workflow Visualization: From BAM to Statistical Readiness
Title: Workflow for Gene Count Matrix Creation
5. The Scientist's Toolkit: Essential Reagents & Resources
Table 2: Key Research Reagent Solutions for Count Matrix Generation
| Item | Function & Relevance |
|---|---|
| Subread/featureCounts | Core software package for efficient read summarization into genomic features. Essential for alignment-based counting. |
| ENSEMBL/UCSC GTF File | High-quality, version-controlled annotation file defining genomic feature coordinates and relationships. Critical for accurate read assignment. |
| R/Bioconductor (DESeq2, tximport) | Statistical environment and packages for importing, processing, and analyzing the count matrix. The primary destination for the matrix. |
| SAMtools | Utilities for handling BAM files (sorting, indexing, querying). Often a prerequisite for counting. |
| Multi-threaded HPC/Cloud Environment | Provides the computational resources necessary for rapid parallel processing of multiple high-coverage BAM files. |
| RNA-seq Alignment Software (STAR, HISAT2) | Generates the input BAM files. Alignment quality directly impacts count accuracy. |
Within a broader thesis on creating a gene count matrix from BAM files, understanding the specific file formats that constitute the workflow is fundamental. This Application Note details the core data formats—SAM, BAM, GTF/GFF3, and the final count table—their roles, structures, and interconversions, providing protocols for their handling in RNA-seq analysis.
Core File Formats: Structures and Comparisons
SAM (Sequence Alignment/Map)
A plain-text, human-readable format storing aligned sequencing reads relative to a reference genome. Each line represents a single read alignment, with 11 mandatory tab-delimited fields.
Table 1: Mandatory SAM Fields (Fields 1-11)
| Field | Name | Description | Example |
|---|---|---|---|
| 1 | QNAME | Query template name | read_12345 |
| 2 | FLAG | Bitwise flag (interpretation of alignment) | 99 |
| 3 | RNAME | Reference sequence name | chr1 |
| 4 | POS | 1-based leftmost mapping position | 10000 |
| 5 | MAPQ | Mapping Quality (Phred-scaled) | 60 |
| 6 | CIGAR | CIGAR string (alignment details) | 50M3I47M |
| 7 | RNEXT | Reference name of mate/next read | chr1 |
| 8 | PNEXT | Position of mate/next read | 10100 |
| 9 | TLEN | Observed template length | 150 |
| 10 | SEQ | Read sequence | ATCGATCG... |
| 11 | QUAL | Read quality (ASCII-encoded Phred scores) | JJJJJ... |
BAM (Binary Alignment/Map)
The compressed, indexed, binary equivalent of SAM. It is not human-readable but is efficient for storage and computational access, making it the de facto standard for downstream analysis.
Table 2: SAM vs. BAM Comparison
| Feature | SAM | BAM |
|---|---|---|
| Format | Plain text | Binary (compressed) |
| Size | Large (~10-30x BAM) | Small (compressed) |
| Human-readable | Yes | No |
| Indexable | No | Yes (.bai file) |
| Primary Use | Inspection, Interchange | Storage & Analysis |
Protocol 1.1: Converting SAM to BAM and Indexing Objective: Convert a SAM file to a sorted, indexed BAM file for efficient downstream processing. Materials: SAM file, reference genome, SAMtools software. Procedure:
- Sort and Convert: Use
samtools sortto sort alignments by genomic coordinate and output as BAM.samtools sort -o aligned.sorted.bam aligned.sam - Index: Generate a BAM index (.bai) for rapid random access.
samtools index aligned.sorted.bam - Validation: Check BAM file integrity.
samtools quickcheck aligned.sorted.bam
GTF (Gene Transfer Format) & GFF3 (General Feature Format version 3)
These are annotation files that describe the coordinates and structure of genomic features (genes, exons, transcripts).
Table 3: GTF/GFF3 Feature Comparison
| Aspect | GTF (v2.2) | GFF3 |
|---|---|---|
| Key Fields | 9 tab-delimited: seqname, source, feature, start, end, score, strand, frame, attributes | 9 tab-delimited (same) |
| Attribute Separator | Semicolon (;) |
Semicolon (;) |
| Key-Value Separator | Space | Equals sign (=) |
| Parent-Child Linking | Uses transcript_id, gene_id |
Uses ID and Parent attributes |
| Feature Hierarchy | Implicit | Explicit |
Protocol 1.2: Preparing a GTF File for Read Counting Objective: Ensure the GTF annotation file is correctly formatted and compatible with featureCounts or HTSeq. Materials: Reference GTF file from Ensembl or GENCODE. Procedure:
- Inspect: View the first few lines to confirm format.
head -5 annotation.gtf - Validate: Check for required attributes (
gene_id,transcript_idfor GTF). - Sort (if required): Some tools require a sorted GTF file.
sort -k1,1 -k4,4n annotation.gtf > annotation.sorted.gtf
From BAM to Count Table: The Core Workflow
The generation of a gene count matrix involves assigning aligned reads (BAM) to genomic features defined in an annotation file (GTF/GFF3).
Diagram 1: RNA-seq Gene Count Matrix Workflow.
The Final Count Table
The output is a tabular file where rows represent features (genes), columns represent samples, and values are integer read counts.
Table 4: Structure of a Final Gene Count Table
| GeneID | Sample1Counts | Sample2Counts | Sample3Counts | ... | Description |
|---|---|---|---|---|---|
| ENSG00000123456 | 150 | 210 | 95 | ... | Gene Symbol A |
| ENSG00000765432 | 0 | 5 | 0 | ... | Gene Symbol B |
| ENSG00000543210 | 2845 | 3012 | 2950 | ... | Gene Symbol C |
Protocol 2.1: Generating a Count Table using featureCounts Objective: Quantify gene-level reads from a BAM file using a GTF annotation. Materials: Sorted BAM file, corresponding BAM index (.bai), GTF annotation file, featureCounts (from Subread package). Procedure:
- Basic Command: Run
featureCountsin gene-assignment mode.featureCounts -T 8 -a annotation.gtf -o counts.txt aligned.sorted.bam-T 8: Use 8 CPU threads.-a annotation.gtf: Path to GTF file.-o counts.txt: Output file.
- Strandedness: Specify library preparation protocol with
-sflag.-s 1for stranded,-s 2for reverse-stranded,-s 0for unstranded. - Output: The main output
counts.txtcontains the count matrix. Thecounts.txt.summaryfile contains assignment statistics.
Protocol 2.2: Aggregating Multiple Samples into a Count Matrix
Objective: Merge individual featureCounts outputs into a unified matrix for differential expression analysis.
Materials: Count files from multiple samples (e.g., sample1.counts, sample2.counts), R/Python environment.
Procedure in R:
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 5: Key Reagents and Tools for BAM-to-Count Matrix Analysis
| Item | Function/Brand Example | Brief Explanation of Role |
|---|---|---|
| Alignment Software | STAR, HISAT2 | Maps RNA-seq reads from FASTQ to reference genome, outputting SAM/BAM. |
| SAM/BAM Utilities | SAMtools, Picard Tools | For format conversion, sorting, indexing, and QC of alignment files. |
| Read Counting Tool | featureCounts (Subread), HTSeq | Quantifies reads overlapping genomic features defined in GTF/GFF3. |
| Annotation Database | ENSEMBL, GENCODE, RefSeq | Provides high-quality, curated GTF/GFF3 files for reference genomes. |
| High-Performance Computing | Cluster/Slurm, Cloud (AWS/GCP) | Provides computational resources for memory/intensive alignment steps. |
| Differential Expression Suite | DESeq2, edgeR (R/Bioconductor) | Consumes the final count matrix for statistical analysis of gene expression. |
Within the context of a thesis on generating a gene count matrix from BAM files, the selection and mastery of specific command-line tools are critical. The process transforms raw sequencing alignment data (BAM files) into a quantitative gene expression matrix, which serves as the fundamental input for downstream differential expression analysis. This application note details the protocols for using SAMtools, Subread (featureCounts), and HTSeq, which represent the core ecosystem for this task. The prerequisite knowledge includes familiarity with Unix/Linux command line, RNA-seq fundamentals, and the general feature file format (GTF/GFF).
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details the essential software "reagents" required for creating a gene count matrix from BAM files.
Table 1: Key Research Reagent Solutions for Gene Count Matrix Generation
| Item Name | Function & Purpose | Key Parameters/Notes |
|---|---|---|
| SAMtools | A suite of utilities for manipulating alignments in SAM/BAM format. Used for sorting, indexing, and filtering BAM files, which is a prerequisite for most counting tools. | samtools sort, samtools index, samtools view -q (MAPQ filtering). |
| Subread (featureCounts) | A highly efficient and feature-rich read summarization program. It assigns aligned reads to genomic features (e.g., genes) and is optimized for speed and memory usage. | -p (paired-end), -t (feature type, e.g., exon), -g (attribute type, e.g., gene_id), -s (strand specificity). |
| HTSeq (htseq-count) | A Python-based script for counting reads overlapping with features. Offers precise handling of ambiguous reads and is widely used in legacy pipelines. | -f bam, -s reverse/yes/no, -t (feature type), -i (ID attribute). Mode (--mode) is critical (e.g., union). |
| Reference Genome | The FASTA file of the reference genome sequence to which reads were aligned. Required for some advanced normalization steps. | Typically from Ensembl, GENCODE, or UCSC. |
| Gene Annotation File | A GTF or GFF3 file defining the coordinates and metadata of genomic features (genes, exons, transcripts). | Must match the reference genome build. Key attributes: gene_id, gene_name. |
| BAM File(s) | The binary alignment files from an aligner like STAR, HISAT2, or TopHat2. The primary input for the counting tools. | Must be coordinate-sorted and indexed for efficient access. |
Experimental Protocols
Protocol 3.1: BAM File Preprocessing with SAMtools
Objective: Prepare coordinate-sorted, indexed BAM files for read counting.
Input: One or more BAM files from a RNA-seq aligner (may be unsorted).
Output: Coordinate-sorted BAM files with accompanying .bai index files.
Methodology:
- Sort by Coordinate: Use
samtools sortto sort alignments by their reference position. This is mandatory for most counters. - Index the Sorted BAM: Create an index file for fast random access.
- Optional Quality Filtering: Filter reads based on mapping quality (MAPQ) to remove poorly aligned reads. A common threshold is MAPQ > 10. Re-index the filtered BAM file if this step is performed.
Protocol 3.2: Read Counting with Subread's featureCounts
Objective: Generate raw gene-level counts from sorted BAM files efficiently. Input: Coordinate-sorted BAM file(s), GTF annotation file. Output: A single text file containing a count matrix for all samples.
Methodology:
- Single Sample Counting:
-s 2: For reverse-stranded libraries (common in Illumina TruSeq). Use-s 1for forward-stranded,-s 0for unstranded.-p: Use this flag if the data is paired-end.
- Multi-Sample Counting (Recommended for consistency): Process all samples simultaneously to get a unified matrix.
- Output: The file
all_samples.counts.txtcontains the raw count matrix. The*.summaryfile contains assignment statistics.
Protocol 3.3: Read Counting with HTSeq-count
Objective: Generate raw gene-level counts using the precise overlap rules of HTSeq. Input: Coordinate-sorted BAM file, GTF annotation file. Output: A simple two-column file (gene_id, count) for one sample.
Methodology:
- Run htseq-count: The primary command for counting. Strandness (
-s) and counting mode (--mode) are crucial.--mode union: This is the most commonly used mode. Reads are counted if they overlap any exon of a gene.-s reverse: For reverse-stranded libraries.
- Multi-Sample Processing: HTSeq does not natively produce a matrix. A wrapper script (e.g., in Bash or Python) is required to run it on each sample and merge the results.
- Handling Ambiguity: The final lines of the output report counts for ambiguous, nofeature, toolow_aQual, etc. These should be excluded from the final count matrix.
Table 2: Performance and Output Comparison of Counting Tools (Typical Values)
| Metric | Subread (featureCounts) | HTSeq-count | Notes / Context |
|---|---|---|---|
| Speed (per sample) | ~5-15 minutes | ~30-90 minutes | For a 30M read mammalian sample. featureCounts is significantly faster. |
| Memory Usage | Low (~4-8 GB) | Moderate (~4-8 GB) | Both are efficient, but featureCounts often uses less. |
| Default Count Mode | "Union" (approximate) | "Union" (strict) | HTSeq's union mode is more conservative in overlapping regions. |
| Paired-end Handling | Built-in (-p) |
Requires -r pos/name |
featureCounts has more straightforward flags. |
| Multi-threading | Yes (-T) |
No | featureCounts can leverage multiple cores for faster processing. |
| Multi-sample Matrix | Direct output | Requires post-processing | featureCounts outputs a combined matrix directly. |
| Ambiguous Read Rule | By fractional count (default) or discard | User-defined mode (--nonunique, etc.) |
featureCounts' fractional counting can be useful for multi-mapping reads. |
Table 3: Typical Read Assignment Statistics from featureCounts Output
| Assignment Status | Read Count (Millions) | Percentage of Total | Interpretation |
|---|---|---|---|
| Assigned | 25.4 M | 84.7% | Successfully assigned to a gene. |
| Unassigned_Ambiguity | 2.1 M | 7.0% | Overlaps multiple genes (depends on tool/mode). |
| Unassigned_NoFeatures | 1.5 M | 5.0% | Does not overlap any annotated gene/exon. |
| Unassigned_TooLowQual | 0.8 M | 2.7% | Fails quality thresholds (e.g., MAPQ). |
| Unassigned_Unmapped | 0.2 M | 0.7% | Read was not aligned. |
| Total | 30.0 M | 100% | Total processed reads. |
Visualization of Workflows
Title: Workflow for gene count matrix generation.
Title: Logic of read assignment in counting tools.
The Practical Workflow: Step-by-Step Guide to Matrix Generation with featureCounts and HTSeq
This protocol constitutes the critical first step in generating a gene count matrix from RNA-seq data, a foundational process for downstream differential expression analysis in drug discovery and biomedical research. Proper preparation and indexing of Binary Alignment/Map (BAM) files ensure data integrity and enable efficient access for read counting tools.
Application Notes
A BAM file is the binary, compressed version of a SAM (Sequence Alignment/Map) file, containing aligned sequencing reads. Indexing creates a companion .bai file, which allows rapid random access to alignments within specific genomic regions without reading the entire file. This is non-negotiable for modern read counters like featureCounts and HTSeq. Skipping or incorrectly performing this step leads to failure in subsequent quantification.
Key Quantitative Metrics for BAM File QC (Pre-Indexing):
| Metric | Ideal Value/Status | Purpose & Interpretation | Common Tool for Assessment |
|---|---|---|---|
| File Integrity | No errors reported | Confirms BAM is not corrupted and can be read. | samtools quickcheck -v |
| Alignment Rate | >70-90% (context-dependent) | Percentage of total reads successfully aligned to the reference genome. | samtools flagstat |
| Properly Paired | High percentage of total pairs | For paired-end data, indicates reads align as correct, oriented pairs. | samtools flagstat |
| Duplicate Rate | Acceptable per experiment | PCR or optical duplicates; may need marking/removal. | samtools markdup / Picard |
| Insert Size | Matches library prep | Checks library preparation quality. | samtools stats / Picard |
| Index Status | .bai file present |
Required for region-based access by counting tools. | Check for .bai file |
Experimental Protocols
Protocol 1: Basic Preparation and Integrity Check of BAM Files
Objective: Validate and prepare a BAM file for indexing and downstream analysis.
Materials:
- SAMtools (v1.15 or later)
- Sorted BAM file (
sample.sorted.bam) - Unix-based command-line environment
Methodology:
- Installation: Ensure SAMtools is installed and in your
PATH. Verify version:samtools --version. - Integrity Check: Validate the BAM file structure. No output indicates a valid file. Any error messages must be addressed.
- Sorting (If Required): Most aligners output sorted BAMs. If yours is unsorted, sort by genomic coordinates:
- Generate Basic Statistics: Obtain alignment summary statistics. Review the output file to assess overall alignment quality.
Protocol 2: Indexing the Sorted BAM File
Objective: Create a BAM index (sample.sorted.bam.bai) to enable fast region-based querying.
Methodology:
- Indexing Command: Execute on the coordinate-sorted BAM file.
This creates
sample.sorted.bam.baiin the same directory. - Verification: Verify the index was created successfully and is compatible. This command will only work with a properly indexed BAM file and outputs contig statistics.
Mandatory Visualization
BAM File Preparation & Indexing Workflow
BAM Index Enables Fast Random Access for Counting
The Scientist's Toolkit
| Research Reagent / Tool | Function in Protocol |
|---|---|
| SAMtools Suite | Core software package for manipulating SAM/BAM files. Provides sort, index, flagstat, and quickcheck. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | BAM file processing is computationally intensive, requiring substantial memory and CPU. |
| Sorted BAM File | Input data. Must be sorted by genomic coordinate (chromosome, then position) for indexing. |
| Reference Genome Index | Used by the aligner to generate the BAM file. Necessary for understanding alignment metrics. |
| QC Report (Flagstat Output) | Text file summarizing alignment statistics to confirm sample quality before proceeding. |
| BAM Index (.bai file) | The binary index file output by samtools index. Essential companion for the sorted BAM. |
| Integrated Development Environment (IDE) / Terminal | Environment for executing command-line steps and managing pipeline scripts. |
Application Notes
Within the thesis workflow "How to create a gene count matrix from BAM files", the step following alignment and file preparation is quantification. The choice of quantification tool is critical, as it determines the type and statistical robustness of the resulting count data. This note compares three seminal tools: featureCounts, HTSeq-count, and Salmon. While featureCounts and HTSeq-count perform alignment-based counting on BAM files, Salmon uses pseudoalignment to estimate transcript abundance from raw reads, which is then often summarized to the gene level.
Quantitative Tool Comparison
Table 1: Core Tool Characteristics & Performance Metrics
| Feature | featureCounts (Subread package) | HTSeq-count | Salmon (quasi-mapping mode) |
|---|---|---|---|
| Primary Method | Alignment-based, direct gene-level counting. | Alignment-based, direct gene-level counting. | Alignment-free, transcript-level quantification via selective-alignment. |
| Input | SAM/BAM files + GTF/GFF annotation. | SAM/BAM files (sorted by name) + GTF/GFF annotation. | Raw FASTQ files or SAM/BAM; transcriptome (FASTA) index. |
| Speed | Very Fast (~5-10x faster than HTSeq). | Slow to Moderate. | Extremely Fast with rich statistical modeling. |
| Multi-mapping Reads | Handles them via --fraction option. |
Default mode discards them (--nonunique none). |
Probabilistically assigns them using an EM algorithm. |
| Strandedness | Explicit parameter (-s 0,1,2). |
Explicit parameter (-s yes,no,reverse). |
Inferred from library type flag (-l). |
| Quantification Level | Primarily gene-level (also exons, etc.). | Primarily gene-level. | Transcript-level, aggregated to gene. |
| Output | Simple count matrix. | Simple count matrix. | Estimated counts (NumReads) & TPM, not raw integers. |
| Key Strength | Speed, efficiency, built-in multi-core support. | Precision, unambiguous counting scheme. | Speed, accuracy, incorporation of sample-specific bias correction (GC, sequence, length). |
Table 2: Practical Implementation Metrics (Typical Human RNA-seq Sample)
| Metric | featureCounts | HTSeq-count | Salmon |
|---|---|---|---|
| RAM Usage | Low (< 4 GB) | Low (< 4 GB) | Moderate (8-16 GB for indexing) |
| CPU Utilization | Excellent parallelization. | Single-core by default. | Excellent parallelization. |
| Data Dependency | Requires final, coordinate-sorted BAM. | Requires name-sorted BAM for paired-end. | Can bypass alignment; works directly with reads. |
| Bias Correction | No. | No. | Yes (sequence, GC, length). |
Experimental Protocols
Protocol 1: Gene-Level Counting with featureCounts Objective: Generate a gene count matrix from a coordinate-sorted BAM file.
- Input Preparation: Ensure BAM files are coordinate-sorted and indexed using
samtools sortandsamtools index. Prepare a comprehensive gene annotation file in GTF format. - Command Execution: Run featureCounts with key parameters.
- Matrix Assembly: The output file
counts.txtcontains a count matrix. The auxiliary filecounts.txt.summaryprovides assignment statistics.
Protocol 2: Gene-Level Counting with HTSeq-count Objective: Precisely assign reads to genes using HTSeq's union-intersection-nonempty mode.
- Input Preparation: For paired-end data, BAM files must be sorted by read name (
samtools sort -n). Prepare a GTF annotation file. - Command Execution: Run htseq-count with appropriate settings.
- Matrix Assembly: Combine individual sample count files (excluding final summary lines) into a single matrix using a scripting language (R, Python).
Protocol 3: Transcript-Level Quantification with Salmon for Gene-Level Analysis Objective: Obtain gene counts via transcript quantification with bias-aware modeling.
- Index Creation: Build a decoy-aware Salmon index for the transcriptome.
- Quantification: Run Salmon in quasi-mapping mode.
- Gene-Level Aggregation: Use the
tximportpackage in R to importquant.sffiles, summarize transcript-level TPM/counts to the gene level, and adjust for average transcript length and count bias.
Visualizations
dot
Tool Selection Workflow for Gene Counting
dot
Tool Inputs and Outputs Comparison
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Digital Tools for RNA-seq Quantification
| Item | Function/Description |
|---|---|
| Coordinate-sorted BAM file | The primary input for alignment-based tools. Contains genomic coordinates of aligned reads. Sorting is required for featureCounts. |
| Name-sorted BAM file | Required for HTSeq-count with paired-end data, ensuring read pairs are processed together. |
| Reference Genome GTF/GFF | Gene annotation file defining genomic coordinates and relationships of exons, transcripts, and genes. Critical for assigning reads. |
| Transcriptome FASTA | A file of all cDNA sequences for the target species. Used by Salmon to build its index for pseudoalignment. |
| Salmon Transcriptome Index | A pre-built, memory-efficient index of the transcriptome, enabling rapid quasi-mapping of reads. |
| High-Performance Computing (HPC) Cluster or Server | Necessary for handling large RNA-seq datasets due to the memory and CPU requirements of alignment and quantification steps. |
| R Statistical Environment with tximport | The software environment used to aggregate Salmon's transcript-level estimates into a stable gene-level count matrix for downstream analysis. |
Within the broader thesis workflow for creating a gene count matrix from RNA-seq BAM files, featureCounts (part of the Subread package) is a critical step for quantifying read alignments against genomic features. It is favored for its efficiency, accuracy, and compatibility with downstream differential expression tools.
Key Command-Line Arguments and Quantitative Comparisons
The utility of featureCounts is governed by its arguments, which control input, annotation, counting modes, and output. The following tables summarize the core arguments and their performance implications.
Table 1: Essential Input/Output Arguments
| Argument | Description | Typical Value | Best Practice Note |
|---|---|---|---|
-a <annotation.gtf> |
Path to GTF/GFF annotation file. | Homo_sapiens.GRCh38.109.gtf |
Use Ensembl/GENCODE annotation matching your reference genome. |
-o <output.path> |
Path for the output count file. | ./counts.txt |
Specify a clear, project-relevant name. |
-s <int> |
Strand specificity. 0 (unstranded), 1 (stranded), 2 (reversely stranded). | 1 or 2 | Must match your library preparation kit. Mis-specification causes major errors. |
-T <int> |
Number of threads. | 4-16 | Increases speed. Do not exceed available cores. |
-p |
Count fragments (reads) instead of reads for paired-end data. | N/A | Use -p -B -C together for paired-end best practices. |
-B |
Only count read pairs where both ends are aligned. | N/A | Recommended for paired-end to improve accuracy. |
-C |
Do not count chimeric/fragments mapping to different chromosomes. | N/A | Recommended to exclude likely artifacts. |
Table 2: Counting Behavior & Performance Arguments
| Argument | Description | Impact on Counts | Performance Impact |
|---|---|---|---|
-t <string> |
Feature type to count (GTF attribute). | exon |
Default (exon) is standard for gene-level counts. |
-g <string> |
Attribute used to group features (e.g., exons into genes). | gene_id |
Must match identifier in GTF attribute column. |
-O |
Assign reads to all overlapping meta-features (genes). | Increases counts for ambiguous reads. | Moderate slowdown. |
--minOverlap <int> |
Minimum overlap required for assignment. | Default 1. Increasing reduces spurious assignment. | Negligible. |
--primary |
Count only primary alignments. | Excludes multi-mapping reads. Recommended. | Reduces downstream noise. |
-M |
Count multi-mapping reads. | Increases counts, adds ambiguity. | Not recommended for standard gene counts. |
-Q <int> |
Minimum mapping quality score. | 10-30 | Filter out low-confidence alignments. |
Detailed Experimental Protocol: Executing featureCounts for Gene-Level Counts
Protocol: Generating a Gene Count Matrix from Multiple BAM Files
Objective: To quantify aligned reads from multiple RNA-seq samples at the gene level using featureCounts, generating a count matrix suitable for downstream analysis (e.g., DESeq2, edgeR).
Materials:
- Subread package (featureCounts) installed (v2.0.6 or higher).
- Sorted BAM files from alignment (e.g., STAR, HISAT2).
- Corresponding genome annotation file (GTF format).
- High-performance computing cluster or workstation with multiple CPU cores.
Procedure:
- Preparation: Organize your directory. Ensure all BAM files and the annotation GTF file are accessible. Verify the strandness (
-s) of your library prep. - Base Command Construction: For a single-sample, paired-end, stranded experiment:
- Batch Processing: To process multiple samples efficiently and create a unified matrix, use a loop or provide all BAM files at once:
- Output Interpretation: The main output file (
all_samples.counts.txt) is a tab-delimited table. The first six columns contain gene identifiers, lengths, etc. Columns 7 onward contain the raw counts per sample. A separate*.summaryfile contains assignment statistics. - Matrix Preparation for Downstream Analysis: The count matrix must be extracted, often by removing the first six columns, and transposed to have genes as rows and samples as columns, as required by most differential expression packages.
Visualizations
Diagram 1: featureCounts Workflow in Gene Matrix Creation
Diagram 2: Read Assignment Logic by featureCounts
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for featureCounts Execution
| Item | Function in Protocol | Example/Note |
|---|---|---|
| Subread/featureCounts Software | Core program for read quantification. | Install via conda (conda install -c bioconda subread) or compile from source. |
| Genome Annotation File (GTF) | Provides coordinates of genomic features (genes, exons). | Ensembl, GENCODE, or RefSeq. Must match reference genome build used for alignment. |
| High-Quality Aligned BAM Files | Input data containing mapped sequencing reads. | Should be sorted, ideally duplicate-marked. Generated by aligners like STAR or HISAT2. |
| High-Performance Compute (HPC) Resource | Enables parallel processing (-T argument) for speed. |
Cluster node or multi-core workstation (8-32 cores recommended). |
| Library Preparation Kit Documentation | Determines the critical -s (strandness) parameter. |
Illumina TruSeq Stranded mRNA: use -s 2. Know your kit's strand specificity. |
| Downstream Analysis Package | Defines final count matrix format requirements. | DESeq2, edgeR, or limma-voom. Guides the final formatting step of the count table. |
Application Notes
HTSeq-count is a pivotal tool in RNA-seq analysis for quantifying gene expression by assigning aligned reads to genomic features. The accuracy of the resulting gene count matrix is highly dependent on the correct specification of the library preparation strandedness parameter (--stranded). This step is critical for downstream differential expression analysis in drug target discovery and biomarker identification.
The --stranded parameter accepts three values: yes, no, and reverse. The choice is dictated by the library construction protocol, which determines the orientation of the sequenced RNA fragment relative to the original mRNA transcript.
Table 1: HTSeq-count --stranded Parameter Settings and Common Library Kits
--stranded Value |
Read Alignment to Coding Strand | Typical Library Kit | Recommended Use Case |
|---|---|---|---|
no (default) |
Ignored. A read is counted if it overlaps a feature, regardless of strand. | Standard, non-stranded kits (e.g., TruSeq Non-stranded). | Historical data or protocols where RNA is not selectively converted to cDNA from the first strand. |
yes |
The read must map to the same strand as the feature (gene). | Stranded kits where the first cDNA strand is synthesized from the original RNA strand (e.g., dUTP method, Illumina TruSeq Stranded kits). | Standard Illumina TruSeq Stranded mRNA, most modern RNA-seq studies. |
reverse |
The read must map to the strand opposite to the feature. | Stranded kits where the second cDNA strand is synthesized from the original RNA strand (e.g., ScriptSeq). | Certain directional kits like Illumina ScriptSeq v2, NuGEN Ovation. |
Table 2: Impact of Incorrect Strandedness Specification (Simulated Data Example)
| Gene ID | True Count (--stranded yes) |
Miscounted (--stranded no) |
Error (%) |
|---|---|---|---|
| GeneA (Sense) | 150 | 205 | +36.7 |
| GeneB (Antisense) | 30 | 75 | +150.0 |
| GeneC (Overlap) | 100 | 100 | 0.0 |
Experimental Protocols
Protocol 1: Determining Library Strandedness Experimentally
Purpose: To empirically determine the correct --stranded parameter for HTSeq-count when library preparation metadata is unavailable.
Materials:
- Aligned BAM file from RNA-seq experiment.
- Reference genome GTF annotation file.
- Known positively expressed, strand-specific gene set (e.g., mitochondrial genes, highly expressed tissue-specific genes).
Methodology:
- Run HTSeq-count in intersection-nonempty mode on a subset of chromosomes for all three strandedness options:
- Identify a control gene known to be transcribed from a specific strand (e.g., MT-ND1 is on the mitochondrial heavy strand).
- Compare counts across the three outputs. The parameter yielding the highest count for the control gene is typically correct, as it correctly assigns reads mapping to the gene's strand.
- Validate with multiple control genes from both positive and negative strands.
Protocol 2: Generating the Final Gene Count Matrix
Purpose: To execute HTSeq-count across multiple samples for consolidated matrix creation. Methodology:
- Parameter Finalization: Confirm the
--strandedparameter (e.g.,yes) using Protocol 1 or kit documentation. - Batch Script Execution: Run HTSeq-count uniformly on all sample BAM files.
- Matrix Compilation: Use a scripting language (Python/R) to merge individual count files, using gene IDs as rows and sample names as columns. Discard summary lines (ending with
__). - Quality Control: Check that counts for housekeeping genes (e.g., ACTB, GAPDH) are present and non-zero across samples.
Visualizations
HTSeq-count Workflow for Count Matrix
Library Types & HTSeq-count Strandedness
The Scientist's Toolkit
Table 3: Research Reagent Solutions for RNA-seq & HTSeq-count
| Item | Function/Description |
|---|---|
| Stranded mRNA Library Prep Kit (e.g., Illumina TruSeq Stranded mRNA) | Provides all reagents for poly-A selection, stranded cDNA synthesis, adapter ligation, and indexing. The specific chemistry determines the --stranded parameter. |
| Alignment Software (e.g., STAR, HISAT2) | Generates the BAM input file for HTSeq-count. Must be run with correct strandedness-aware parameters to preserve compatibility. |
| High-Quality Reference GTF (e.g., from Ensembl, GENCODE) | Contains gene model annotations (exon boundaries, gene IDs, strand). Must match the genome build used for alignment. |
| HTSeq-count Software (v2.0.3+) | Python package for parsing BAM and GTF files to produce raw gene counts. The core tool for the quantification step. |
| SAMtools | Utilities for manipulating BAM files (sorting, indexing) which may be a prerequisite for HTSeq-count. |
| Computational Environment (e.g., Linux server, HPC, Python environment) | Essential for running memory- and CPU-intensive tasks of alignment and read counting. |
Application Notes
In constructing a gene count matrix from RNA-seq BAM files, a critical challenge is the accurate assignment of sequencing reads that map to multiple genomic locations (multi-mapping reads) or that overlap multiple genomic features (e.g., exon boundaries of different genes). Improper handling leads to quantification bias, compromising downstream differential expression analysis. Current best practices involve probabilistic assignment using expectation-maximization algorithms or weighted distribution schemes, implemented in tools like featureCounts, Salmon, and RSEM.
Key Quantitative Considerations:
- Prevalence: Approximately 5-20% of reads in a standard RNA-seq experiment are multi-mapping, with higher rates in genomes with high sequence similarity (e.g., paralogous genes).
- Assignment Impact: Naïve exclusion of multi-mapping reads can reduce power and introduce false negatives, while simple random assignment can increase false positives.
- Performance Metrics: Modern tools achieve >95% accuracy in simulated benchmark datasets for unambiguous reads, but accuracy drops to 70-85% for highly repetitive regions, emphasizing the need for curated annotation.
Table 1: Comparison of Primary Tools for Handling Ambiguity
| Tool | Primary Strategy for Multi-mapping Reads | Handling of Overlapping Features | Recommended Use Case | Reported Assignment Accuracy* |
|---|---|---|---|---|
| featureCounts | Uniform weighting or exclusion. | Counts to all overlapping features or a single feature. | Simple, fast gene-level counting. | ~85% (with –M –fraction) |
| Salmon | Expectation-Maximization (EM) with transcript-aware mapping. | Probabilistic resolution at transcript level. | Accurate transcript-level quantification. | ~92% |
| STAR | Unique mapping only or random assignment. | Primarily for alignment; counting requires a secondary tool. | Initial alignment and splice junction detection. | N/A (Alignment tool) |
| RSEM | Bayesian probabilistic model with EM. | Models read generation from transcripts. | Detailed transcript isoform quantification. | ~90% |
| HTSeq | Intersection-strict, union, etc. Can discard ambiguous reads. | Rule-based assignment based on overlap mode. | Simple counting with strict gene models. | ~82% (union mode) |
*Accuracy percentages are approximations from published benchmarks on simulated data.
Experimental Protocols
Protocol 1: Probabilistic Assignment with featureCounts (Subread package)
This protocol generates a raw gene count matrix while fractionally assigning multi-mapping reads.
- Input: Coordinate-sorted BAM file(s) from a spliced aligner (e.g., STAR), a Gene Transfer Format (.GTF) annotation file.
- Installation: Install Subread package (
conda install -c bioconda subread). - Command Execution:
- Output Processing: The file
counts.txtcontains a matrix with gene identifiers and raw counts. The summary file contains statistics on assigned/unassigned reads.
Protocol 2: Transcript-Aware Quantification with Salmon
This protocol performs lightweight alignment and quantification directly from raw reads, intrinsically handling multi-mapping via its statistical model.
- Input: Raw FASTQ files, a transcriptome index.
- Index Generation: Download a cDNA reference (e.g., from Ensembl) and build an index:
salmon index -t transcripts.fa -i salmon_index - Quantification:
- Matrix Construction: Use the
tximportR/Bioconductor package to aggregate transcript-level abundance estimates to the gene level, correcting for potential length biases.
Visualization
Title: Decision Workflow for Ambiguous Reads in Gene Counting
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Resources for Gene Count Matrix Generation
| Item | Function & Relevance |
|---|---|
| High-Quality Reference Genome & Annotation (GTF/GFF) | Essential for accurate read assignment. Ensembl/RefSeq annotations provide standardized gene models for defining feature boundaries. |
| Spliced Read Aligner (e.g., STAR, HISAT2) | Aligns RNA-seq reads across splice junctions, generating the input BAM files. Critical for capturing full transcript information. |
| Counting Software Suite (e.g., Subread/featureCounts, HTSeq) | Core tool that processes BAM and GTF files to assign reads to features, implementing rules for ambiguity. |
| Probabilistic Quantification Tool (e.g., Salmon, kallisto) | Provides an alternative, alignment-free or lightweight-alignment approach that models and resolves multi-mapping reads statistically. |
| Computational Environment (Conda/Bioconda) | Enables reproducible installation and version management of complex bioinformatics toolchains. |
| RNA-seq Simulation Software (e.g., polyester, RSEM-sim) | Generates benchmark datasets with known truth for evaluating the accuracy of counting pipelines in handling ambiguity. |
This document details the critical step of merging individual sample count files into a unified gene count matrix, a foundational dataset for downstream differential expression analysis in genomics research.
The table below compares the primary file formats and merging strategies.
Table 1: Input File Formats and Merge Characteristics
| Format | Typical Columns | Merge Key | Primary Challenge | Recommended Tool |
|---|---|---|---|---|
| FeatureCounts Output | GeneID, Chr, Start, End, Strand, Length, Sample.counts | Gene identifier (e.g., GeneID) | Consistent column order across samples | R: tximport or cbind() |
| HTSeq-Count Output | Gene_ID, counts | First column (Gene_ID) | Header/comment lines, non-gene rows | Python: pandas with careful read |
| RSEM .genes.results | geneid, transcriptid(s), expected_count, TPM, FPKM | gene_id | Extracting only the count column | R: tximport |
| Salmon/Sailfish Quant | Name, Length, EffectiveLength, TPM, NumReads | Transcript/Gene identifier | Transcript-to-gene aggregation | R: tximport / tximeta |
Table 2: Consolidated Matrix Structure
| GeneID | Length | Sample1 | Sample2 | Sample3 | ... | SampleN |
|---|---|---|---|---|---|---|
| ENSG00000187634 | 3662 | 150 | 89 | 300 | ... | 421 |
| ENSG00000188976 | 3712 | 0 | 5 | 12 | ... | 7 |
| ENSG00000187961 | 3995 | 45 | 210 | 78 | ... | 133 |
| ... | ... | ... | ... | ... | ... | ... |
Experimental Protocols
Protocol A: Merging in R using base functions andtximport
- Objective: Create a robust count matrix from individual text-based count files (e.g., from featureCounts).
- Materials: List of all sample file paths, consistent gene identifiers.
- Procedure:
- Read and Validate Individual Files:
- Iterative Merge:
- Protocol B: Merging Quantified Transcripts with
tximport(Recommended for pseudo-aligners)- Prepare a Sample Manifest:
- Import and Summarize to Gene-level:
Protocol C: Merging in Python usingpandas
- Objective: Generate a count matrix using Python's data manipulation libraries.
- Procedure:
Visualization of the Merging Workflow
Title: Workflow for merging sample counts into a matrix.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Count Merging
| Item | Function & Purpose |
|---|---|
R tximport / tximeta |
Robust import and summarization of transcript-level quantifications (Salmon, Kallisto) to gene-level counts, correcting for potential changes in gene length. |
Python pandas |
Core library for data manipulation; provides the DataFrame.join() function essential for merging count tables on a shared index (GeneID). |
| Consistent Gene Annotation (GTF/GFF3) | Provides the tx2gene map for transcript-to-gene summarization and ensures identical gene identifiers across all samples. |
| Sample Manifest (CSV/TSV) | A metadata file linking sample names to file paths, crucial for automated, error-free import and correct column labeling. |
| Version-Controlled Script (Rmd/Jupyter) | Reproducible document that records exact parameters, file paths, and code used to generate the final matrix, ensuring auditability. |
| Integrity Check Script | Validates that all input files have the same number and order of genes, preventing silent merge errors that corrupt downstream analysis. |
Solving Common Pitfalls: Troubleshooting and Optimizing Your Count Workflow
Application Note AN-2024-002
1. Introduction Within the broader research thesis "How to create a gene count matrix from BAM files," a critical challenge is the generation of a count matrix with unexpectedly low gene counts. This compromises downstream differential expression analysis. This Application Note details two primary diagnostic foci: the quality of sequence alignment within BAM files and the potential mismatches between alignment coordinates and gene annotation files (GTF/GFF). We present protocols for systematic diagnosis and remediation.
2. Key Causes and Diagnostic Data Primary causes for low count yields fall into two categories, as summarized in Table 1.
Table 1: Primary Causes of Low Count Yields and Diagnostic Indicators
| Category | Specific Cause | Key Diagnostic Indicator(s) | Typical Impact on Counts |
|---|---|---|---|
| Alignment Quality | High multi-mapping rate | >30% of reads marked as secondary/supplementary in FLAG field. | Reads discarded or improperly assigned, reducing usable reads. |
| Low mapping quality (MAPQ) | High proportion of MAPQ < 10 in BAM file. | Reads filtered out during counting, reducing counts. | |
| Poor adapter/quality trimming | High mismatch rate in aligned region; Soft-clipping at read ends > 10%. | Reads misaligned or filtered, reducing mappability. | |
| Annotation Mismatch | Genome Build Incompatibility | Alignment coordinates do not overlap any annotated feature in GTF. | Near-total loss of counts. |
| Transcript Version Discrepancy | Feature counts succeed but gene IDs are missing or unmatched. | Counts generated but cannot be aggregated to correct gene identifier. | |
| Strandedness Mis-specification | For stranded libraries, sense/antisense counts are reversed. | 50-70% reduction in correct strand counts. |
3. Experimental Protocols
Protocol 3.1: Comprehensive BAM File Alignment Quality Assessment
Objective: Quantify alignment metrics that directly impact feature counting. Materials: BAM file(s), Samtools, FastQC (post-alignment). Procedure:
- Generate Alignment Statistics:
samtools flagstat <input.bam> > alignment_flagstat.txtKey metrics: percentage of properly paired reads, secondary/supplementary alignments. - Calculate Mapping Quality Distribution:
samtools view <input.bam> | awk '{print $5}' | sort | uniq -c > mapq_distribution.txtTabulate the count of reads at each MAPQ score. - Inspect Per-Base Alignment Quality (Optional but recommended):
Use Qualimap RNA-seq mode:
qualimap rnaseq -bam <input.bam> -gtf <annotation.gtf> -outdir qualimap_reportReview the "Mapping Quality" and "Insert Size" plots. Interpretation: A high rate of secondary alignments (>30%) or a majority of reads with MAPQ < 10 indicates fundamental alignment issues likely causing low counts.
Protocol 3.2: Annotation Mismatch Detection and Resolution
Objective: Identify and correct discrepancies between BAM coordinate space and the provided annotation file. Materials: BAM file, Annotation file (GTF/GFF), Genome browser (e.g., IGV), Subread/featureCounts or HTSeq. Procedure:
- Pilot Counting with Detailed Summary:
Using featureCounts (from Subread package):
featureCounts -a <annotation.gtf> -o pilot_counts.txt -T 8 --verbose -p --countReadPairs <input.bam>The--verboseflag outputs a stat file summarizing the fraction of reads assigned, unassigned, and reasons for unassignment. - Analyze Unassigned Reads: If a high percentage (>50%) of reads are "Unassigned_NoFeatures," extract a subset of these reads' coordinates and visualize them in IGV alongside the loaded annotation tracks. Check for obvious genome build misalignment.
- Verify Strandedness:
Perform a strandedness check using
infer_experiment.pyfrom the RSeQC package:infer_experiment.py -r <annotation.bed> -i <input.bam>Compare the inferred "Fraction of reads failed to determine" and strand orientation to your library preparation kit's expected strandedness. Resolution: If a genome build mismatch is found, re-align reads to the correct build or obtain a GTF matching your alignment build. If strandedness is mis-specified, correct the-sparameter in your counter.
4. Visualization of Diagnostic Workflow
Diagram Title: Diagnostic Flow for Low RNA-seq Counts
5. The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for Diagnosis and Generation of Gene Count Matrices
| Tool / Reagent | Primary Function | Role in Diagnosing Low Counts |
|---|---|---|
| Samtools | Manipulate and view SAM/BAM files. | Extract alignment statistics (flagstat, MAPQ distribution) to assess quality. |
| Qualimap | Generate quality control metrics for NGS data. | Provides integrated visual reports on alignment quality relative to annotations. |
| RSeQC | RNA-seq specific quality control. | Infers library strandedness experimentally from BAM and BED files. |
| featureCounts (Subread) | Efficient read counting for genomic features. | --verbose output quantifies reasons for read unassignment. |
| Integrative Genomics Viewer (IGV) | Visual exploration of genomic data. | Visually confirm alignment vs. annotation coordinate mismatches. |
| Species-Specific GENCODE/Ensembl GTF | Authoritative gene annotation. | Ground truth for feature boundaries; correct version eliminates mismatch. |
| Adapter Trimming Tool (e.g., Cutadapt) | Remove adapter sequences from reads. | Pre-processing step to improve alignment rate and specificity. |
| Pseudoalignment-based Counter (e.g., Salmon) | Alignment-free quantification. | Alternative approach; can be more robust to sequence polymorphism and quality issues. |
Within the broader workflow of creating a gene count matrix from BAM files, strand-specificity is a critical parameter. It determines whether the aligned reads in a BAM file retain information about their originating DNA strand. Non-strand-specific (unstranded) protocols pool reads from both strands, complicating the accurate assignment of reads to overlapping genes on opposite strands. Strand-specific (stranded) protocols preserve this orientation, vastly improving precision. Incorrectly specifying this parameter during read counting leads to significant quantification errors, directly impacting downstream differential expression analysis in drug target discovery.
Key Library Preparation Protocols and Their Signatures
The strandedness of a BAM file is dictated by the cDNA library preparation kit. The table below summarizes the most common protocols, their key characteristics, and how they manifest in aligned read data.
Table 1: Common Strand-Specific RNA-Seq Library Protocols
| Protocol (Kit Examples) | Strandedness | Key Reverse Transcription Primer | Critical Read Alignment Signature for Forward Strand Gene (++) | Compatibility with Common Quantification Tools |
|---|---|---|---|---|
| dUTP Second Strand (Illumina TruSeq Stranded, NEBNext Ultra II) | Yes (Reverse) | Oligo-dT / Random, incorporates dUTP in second strand. | Read 1 aligns to the opposite (reverse) strand. | featureCounts: -s 2; HTSeq: --stranded=reverse |
| NSR (Illumina Directional) | Yes (Forward) | Adaptor-anchored, no second strand synthesis. | Read 1 aligns to the same (forward) strand. | featureCounts: -s 1; HTSeq: --stranded=yes |
| Standard Non-Stranded | No | Standard primers, both cDNA strands sequenced. | Reads align to both strands. | featureCounts: -s 0; HTSeq: --stranded=no |
| SMARTer-type (Clontech) | Yes (Forward) | Template-switching oligo (TSO) at 5' end of first strand. | Read 1 aligns to the same (forward) strand. | featureCounts: -s 1; HTSeq: --stranded=yes |
Experimental Protocol: Determining Strand-Specificity Empirically
If the library preparation metadata is unavailable, the strandedness must be determined empirically from the BAM file.
Protocol: In Silico Strandedness Assay
Objective: To unambiguously determine the strandedness parameter (0, 1, or 2) for an RNA-seq BAM file.
Materials & Reagents:
- Input: Coordinate-sorted BAM file from a strand-aware aligner (e.g., HISAT2, STAR).
- Software:
- SAMtools (v1.15+): For BAM file processing and indexing.
- IGV (Integrative Genomics Viewer) or command-line utilities (
bedtools). - R/Bioconductor with
GenomicAlignmentspackage (optional).
Procedure:
- Select Diagnostic Genomic Regions: Identify 20-30 genes with unambiguous, non-overlapping transcription. Ideal candidates are:
- Protein-coding genes with clear strand orientation.
- Genes located on the opposite strand of a known non-coding RNA (e.g., a miRNA host gene).
- Mitochondrial genes or highly expressed, single-isoform genes.
- Visual Inspection in IGV:
- Load the BAM file and its index into IGV.
- Navigate to a selected diagnostic gene (e.g., ACTB on the forward strand).
- Observe Read Alignment Pattern: In the track panel, note the directionality of read pairs.
- If the gene is on the forward strand (++), and Read 1 aligns predominantly to the reverse (-) strand, the library is reverse-stranded (dUTP method,
-s 2). - If Read 1 aligns predominantly to the forward (+) strand, the library is forward-stranded (NSR/SMARTer,
-s 1). - If reads align evenly to both strands, the library is unstranded (
-s 0).
- If the gene is on the forward strand (++), and Read 1 aligns predominantly to the reverse (-) strand, the library is reverse-stranded (dUTP method,
- Quantitative Validation with Feature Counts:
- Using a subset of diagnostic regions in a BED/GTF file, run a counting tool (e.g.,
featureCounts) with all three strandedness parameters (-s 0,-s 1,-s 2). - For a forward-strand gene, the correct parameter will yield >95% of total counts, while the incorrect stranded parameter will yield <5%. The unstranded parameter will yield ~50%.
- Using a subset of diagnostic regions in a BED/GTF file, run a counting tool (e.g.,
- Decision: The parameter yielding >95% of counts for genes of known orientation is the correct
-svalue for the entire dataset.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Tools for Strand-Specific Analysis
| Item | Function in Resolving Strand-Specificity |
|---|---|
| Stranded mRNA Library Prep Kit (e.g., Illumina TruSeq Stranded mRNA) | Generates libraries where the strand-of-origin is encoded via dUTP incorporation, making downstream analysis unambiguous. |
| Ribo-Zero/RiboCop Kits | Deplete ribosomal RNA without biasing strand orientation, preserving strand information in total RNA protocols. |
| Strand-Aware Aligner (STAR, HISAT2, TopHat2) | Aligns reads while considering the splicing signal and correctly interpreting the orientation of paired-end reads relative to the transcript. |
| SAMtools | Indexes and manipulates BAM files, allowing for efficient extraction of reads from diagnostic regions for empirical testing. |
| Strand-Specific Genome Annotation (GTF/GFF) | Contains the "strand" attribute for each feature. Essential for all counting software (featureCounts, HTSeq-count) to assign reads correctly. |
| Diagnostic Region BED File | A custom list of genomic coordinates for high-confidence, strand-unambiguous genes. Used for the empirical strandedness assay protocol. |
Workflow and Pathway Diagrams
Diagram 1: Strand-specific gene count matrix workflow
Diagram 2: Stranded protocol read alignment signatures
Application Notes and Protocols for Gene Count Matrix Generation from BAM Files
This document details optimized protocols for generating a gene count matrix from aligned sequencing data (BAM files), a critical step in RNA-seq analysis pipelines for drug target discovery and biomarker identification. The focus is on overcoming computational bottlenecks in I/O operations and counting processes through parallelization and memory-efficient file handling.
Key Performance Data
Quantitative benchmarks for common tools highlight the trade-offs between speed, memory, and accuracy. Data is synthesized from recent community benchmarks (2023-2024).
Table 1: Comparison of Gene Counting Tools & Strategies
| Tool / Method | Parallelization Strategy | Peak Memory Usage (GB)* | Time for 100 BAM files* | Primary File Handling | Key Advantage |
|---|---|---|---|---|---|
| HTSeq (single) | None (sequential) | 2-4 | 18-24 hours | Iterative BAM read | Baseline, simple logic |
| HTSeq (pool) | Multi-process (BAM file) | 4 x N processes | 4-6 hours | File-level parallel | Easy Python integration |
| featureCounts | Multi-thread (read level) | 5-8 | 2-3 hours | Blockwise BAM parsing | Speed & built-in threading |
| STAR --quantMode | Integrated with alignment | 28-32 | (Included in align) | Streaming SAM/BAM | All-in-one alignment/counting |
| UMI-tools count | Multi-process (cell/barcode) | 6-10 | 6-8 hours | Deduplication-aware | Single-cell UMI handling |
| Python Dask | Cluster-level (chunk BAM) | Configurable | ~1 hour (on cluster) | Chunked BAM files | Extreme scalability |
*Example data based on human transcriptome (60M paired-end reads per sample). Actual values depend on hardware and BAM size.
Core Experimental Protocols
Protocol 3.1: Parallelized Counting with featureCounts (Subread package)
Objective: Utilize multi-threading for fast read assignment to genomic features. Materials: High-performance computing node with ≥8 cores, 16GB RAM, sorted BAM files, GTF annotation file.
- Preparation: Ensure BAM files are coordinate-sorted and indexed (
*.baifiles exist). - Command:
- Post-processing: Extract the count matrix from
gene_counts.txt, removing header and statistical columns.
Protocol 3.2: Memory-Efficient BAM Processing with Sambamba
Objective: Rapidly subset or filter BAM files using multi-threaded, memory-optimized tools to prepare for counting. Materials: Sambamba tool installed, BAM files.
- Filter for uniquely mapped reads (high mapping quality):
- Sort by read name (required by some counters like HTSeq):
Protocol 3.3: Chunked BAM Processing with Python & Pysam
Objective: Process extremely large BAM files in fixed memory by reading in genomic chunks. Materials: Python 3.9+, Pysam library, interval list file.
- Script Workflow:
- Implementation Note: Requires careful merging of results from overlapping genomic features across chunks.
Visualized Workflows
Diagram 1: Parallel Gene Count Matrix Workflow
Diagram 2: Memory-Efficient Chunked Processing Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools & Resources
| Item | Function/Benefit | Typical Use Case |
|---|---|---|
| Sambamba | Faster, multi-threaded alternative to SAMtools for BAM operations. | Filtering, sorting, and indexing BAM files pre-counting. |
| Subread (featureCounts) | Highly optimized and threaded read summarization program. | Primary tool for fast gene-level counting from multiple BAMs. |
| Pysam | Python interface for reading/writing SAM/BAM files. | Custom scripting for complex counting logic or chunked processing. |
| Dask / Snakemake | Frameworks for parallel workflow management on clusters. | Orchestrating massive, scalable counting pipelines across hundreds of samples. |
| Sorted & Indexed BAM | Coordinate-sorted BAM with .bai index enables fast random access. |
Prerequisite for all region-based counting and chunking strategies. |
| High-IOPS Storage | Storage with high Input/Output Operations Per Second (e.g., SSD, NVMe). | Drastically reduces time spent reading/writing intermediate BAM files. |
| GTF/GFF3 Annotation | File specifying genomic coordinates of exons and genes. | Reference for assigning reads to genomic features. Must match reference genome. |
Within the broader thesis on generating a gene count matrix from BAM files, a critical computational challenge is the accurate assignment of sequencing reads that map to multiple genomic locations or to regions shared by multiple gene isoforms. These ambiguous reads, if not handled correctly, lead to quantification inaccuracies that propagate through downstream differential expression and biomarker discovery analyses, directly impacting drug development research. This Application Note details current strategies and protocols for managing such ambiguity.
Quantification of the Ambiguity Problem
The prevalence of ambiguous reads varies significantly by organism and transcriptome complexity.
Table 1: Sources and Estimated Prevalence of Ambiguous Reads in RNA-Seq
| Ambiguity Source | Description | Estimated % of Total Reads (Human) |
|---|---|---|
| Multi-mapped Reads | Reads mapping identically to multiple loci (e.g., paralogous genes, repeats). | 10-30% |
| Overlapping Gene Loci | Reads mapping to genomic regions where gene annotations overlap on the same or opposite strands. | 5-15% |
| Alternative Isoforms | Reads mapping to exon regions shared between different transcripts of the same gene. | 30-60% of gene's reads |
Core Strategies and Methodological Frameworks
Discardment vs. Proportional Assignment
The most basic strategy is to discard all multi-mapped reads, ensuring uniqueness but losing a substantial fraction of data. The preferred standard is proportional assignment, where a read’s count is fractionally distributed among all its candidate alignments based on probabilistic models.
Transcript-Aware Quantification
For isoform ambiguity, tools use transcriptome reference rather than a collapsed gene reference. They model the likelihood of observing a read given the relative abundance of each transcript, considering the specific exon-intron structure.
Resolving Multi-mapping Reads with EM Algorithms
Tools like Salmon and kallisto employ expectation-maximization (EM) algorithms to resolve ambiguity. They initially assign reads fractionally, estimate transcript abundances, and iteratively refine these estimates and read assignments.
Table 2: Comparison of Primary Tools for Handling Ambiguous Reads
| Tool | Algorithm Core | Handles Multi-mapping? | Handles Isoform Ambiguity? | Primary Output |
|---|---|---|---|---|
| featureCounts | Read counting to genomic features. | No (excludes multi-mappers by default). | No (requires gene-level annotation). | Gene Counts |
| Salmon | Lightweight alignment & probabilistic modeling. | Yes, via EM. | Yes, explicitly models transcripts. | Transcript Counts |
| kallisto | Pseudoalignment & EM. | Yes, via pseudoalignment equivalence classes. | Yes, core function. | Transcript Counts |
| STAR | Spliced aligner + built-in quant. | Can output multi-mapped reads flagged. | Limited (via --quantMode). |
Gene/Transcript Counts |
| RSEM | EM after alignment. | Yes, using posterior probabilities. | Yes, core function. | Transcript Counts |
Detailed Protocols
Protocol 1: Transcript-Level Quantification with Salmon (Alignment-Based Mode)
This protocol generates transcript abundance estimates, properly distributing reads across isoforms, from a BAM file.
Materials:
- Sorted BAM file from spliced-aware aligner (e.g., STAR, HISAT2).
- Transcriptome FASTA file (e.g.,
gencode.vXX.transcripts.fa). - Salmon software (v1.9.0 or higher).
- Compute environment with ≥ 16GB RAM.
Procedure:
- Generate Transcript-to-Gene Map: Extract from GTF annotation.
- Prepare Salmon Index:
- Run Salmon Quantification on BAM:
-l A: Automatically infer library type.--gcBias: Correct for GC content bias.- The tool internally applies an EM algorithm to resolve isoform ambiguity.
- Aggregate to Gene Level (if needed): Use the
tximportR/Bioconductor package to summarize transcript-level counts and abundances to the gene level, which is more stable for differential expression.
Protocol 2: Resolving Multi-mapped Reads with STAR & RSEM
A pipeline combining STAR's sensitive alignment with RSEM's rigorous statistical model for multi-mapping and isoform resolution.
Procedure:
- Align with STAR in Transcriptome-aware Mode:
This produces a
TranscriptomeSAMBAM file aligned to the transcriptome. - Quantify with RSEM: RSEM processes the BAM, calculates posterior probabilities for each read alignment, and outputs expected counts for genes and isoforms.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Ambiguity-Resolving Quantification
| Item | Function & Relevance |
|---|---|
| High-Quality, Strand-Specific RNA-Seq Library Prep Kit (e.g., Illumina Stranded mRNA Prep) | Preserves strand information, critical for resolving overlaps on opposite strands and accurate transcript assignment. |
| Spliced Transcriptome Alignment Software (e.g., STAR, HISAT2) | Produces BAM files with splice-aware alignments, the essential input for downstream ambiguity resolution. |
| Probabilistic Quantification Software (e.g., Salmon, kallisto, RSEM) | Core tool that implements mathematical models (EM) to fractionally assign ambiguous reads. |
| Comprehensive & Current Annotation File (e.g., GENCODE, RefSeq GTF) | Defines gene and transcript models; accuracy is paramount. Must match the reference used for alignment. |
| Tximport R/Bioconductor Package | Converts transcript-level abundance estimates from probabilistic tools into a gene-level count matrix suitable for DESeq2/edgeR. |
Visualizations
Title: Workflow Comparison for Ambiguous Read Handling
Title: Probabilistic Assignment of a Read to Two Isoforms
Application Notes
Within the thesis on generating a gene count matrix from BAM files, maintaining data integrity through rigorous Quality Control (QC) is paramount. MultiQC aggregates results from bioinformatics tools across numerous samples into a single interactive report, enabling systematic auditing of count generation steps. This is critical for researchers and drug development professionals who require reproducible, high-quality count matrices for downstream differential expression analysis.
Core Quantitative Metrics for Auditing Count Generation:
| QC Checkpoint Stage | Key Tool | Primary Metric(s) Audited | Acceptable Range/Indicator |
|---|---|---|---|
| Alignment & BAM QC | SAMtools / qualimap | % Aligned Reads, Duplication Rate, Insert Size | >70-80% aligned, Dup rate context-dependent |
| Read Counting | featureCounts / HTSeq | Assigned Reads, Unassigned Reads (No Features, Ambiguous) | High % assigned (>70-80%), low ambiguous |
| Gene/Transcript Level | featureCounts / StringTie | Features (Genes/Transcripts) Detected, Count Distribution | Consistent detection across sample groups |
| Overall Pipeline | MultiQC | Cross-sample consistency of all above metrics | No outliers, uniform distributions |
MultiQC Aggregation Table:
| MultiQC Module | Parses Output From | Visualization in Report | Audit Question Answered |
|---|---|---|---|
| Samtools Stats | samtools stats |
General Stats Table, Sequence Counts | Did alignment yield expected read numbers per sample? |
| Qualimap | Qualimap RNA-seq reports | Genomic Origin, 5'/3' Bias, Coverage | Are alignments properly distributed across features? |
| featureCounts | featureCounts summary log |
Assignment Stat Bar Plot, Count Heatmap | What proportion of reads was counted for genes? |
| Picard MarkDuplicates | Picard metrics files | Duplication Rate Plot | Is duplication level uniform across conditions? |
| FastQC | FastQC zip files | Per-base sequence quality, adapter content | Were initial data quality issues carried forward? |
Detailed Protocols
Protocol 1: Generating Input Files for MultiQC from a Standard Counting Workflow
Objective: Execute a standard RNA-seq alignment and gene counting pipeline while generating tool-specific summary files that MultiQC can parse.
Materials:
- Aligned BAM files (e.g., from HISAT2 or STAR).
- Reference genome annotation file (GTF/GFF).
- Software: SAMtools, Subread (featureCounts), Qualimap, MultiQC.
Methodology:
- BAM File Processing and QC:
Read Counting with FeatureCounts:
Aggregate with MultiQC:
- MultiQC automatically searches for recognizable files (e.g.,
*.log,summary,qualimapReport.html) and compiles them.
- MultiQC automatically searches for recognizable files (e.g.,
Protocol 2: Auditing the Count Matrix Using the MultiQC Report
Objective: Systematically evaluate the quality and consistency of the count generation steps using the generated MultiQC report.
Methodology:
- Open the Report: Launch the
multiqc_report.htmlfile in a web browser. - Audit the "General Statistics" Table:
- Sort columns by "% Aligned", "Assigned", "Duplication Rate".
- Check: Identify any sample with a drastic deviation (>15% difference) from the group median for key metrics. Flag these samples for potential exclusion or re-analysis.
- Investigate the "featureCounts" Section:
- Examine the bar plot for "Assignment status of reads".
- Check: Confirm a high, consistent proportion of "Assigned" reads across all samples. Investigate samples with high "UnassignedNoFeatures" (may indicate contaminant DNA or poor annotation) or high "UnassignedAmbiguous" (may indicate high multi-mapping in repetitive regions).
- Review the "Qualimap" Section:
- Analyze the "Genomic origin of reads" plot. Expect majority from "exonic".
- Check "Cumulative coverage profile" for uniform 5' to 3' coverage.
- Check: Samples with high "intergenic" reads may indicate genomic DNA contamination. Strong 5' or 3' bias may indicate RNA degradation.
- Cross-Correlate with "FastQC" and "Samtools Stats":
- Correlate initial adapter content or poor quality bases with lower alignment and assignment rates.
- Final Audit Decision: Based on convergence of evidence from multiple tools, decide if the count matrix is derived from high-quality, consistent data. Document any excluded samples and the QC rationale.
Visualizations
MultiQC Audit Workflow for Count Matrix
Decision Logic for MultiQC Metric Auditing
The Scientist's Toolkit: Research Reagent Solutions
| Tool / Resource | Category | Primary Function in Auditing Count Generation |
|---|---|---|
| MultiQC | Software | Aggregates results from bioinformatics tools into a single visual report for holistic QC. |
| Subread (featureCounts) | Software | Performs read summarization to genomic features; its summary file provides essential counting statistics. |
| Qualimap | Software | Evaluates alignment characteristics (e.g., genomic origin, bias) in BAM files post-alignment. |
| SAMtools | Software | Provides fundamental utilities (flagstat, stats, idxstats) for processing and checking BAM files. |
| Reference Genome Annotation (GTF/GFF) | Data File | Essential map for assigning reads to features. Quality and version must be consistent with alignment. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | Infrastructure | Provides the computational power to run alignment, counting, and QC tools on large sample sets. |
Ensuring Accuracy: How to Validate and Compare Different Counting Methods
Within the broader thesis research on creating a gene count matrix from BAM files, validation of the resulting expression data is a critical step. This application note details protocols for validating RNA-seq derived gene counts using quantitative PCR (qPCR) correlation and synthetic spike-in controls. These techniques assess technical accuracy, sensitivity, and dynamic range.
Quantitative PCR (qPCR) Correlation Validation
Core Principle
A subset of genes quantified in the gene count matrix is independently measured using qPCR, the gold standard for targeted nucleic acid quantification. A strong correlation (typically Pearson's r > 0.85-0.90) between RNA-seq counts (normalized) and qPCR values (e.g., ∆Ct) supports the accuracy of the bioinformatic pipeline.
Detailed Protocol
Step 1: Gene Selection
- Select 20-50 genes spanning a wide range of expression levels (high, medium, low) from your gene count matrix.
- Include housekeeping genes (e.g., GAPDH, ACTB) and genes relevant to your experimental condition.
- Design and validate qPCR primers/probes for each target. Ensure amplicons are within the exonic regions used for RNA-seq counting.
Step 2: cDNA Synthesis
- Use the same total RNA samples that were submitted for RNA-seq.
- Perform reverse transcription using a high-fidelity kit (e.g., SuperScript IV).
- Use anchored oligo-dT or random hexamers, matching the RNA-seq library preparation protocol.
- Include a no-reverse transcriptase (-RT) control for each sample to detect genomic DNA contamination.
Step 3: Quantitative PCR
- Set up qPCR reactions in triplicate for each gene-sample pair.
- Use a fluorescent dye-based master mix (e.g., SYBR Green) or probe-based assay (e.g., TaqMan).
- Run on a calibrated real-time PCR instrument.
- Include a standard curve (serial dilution of a pooled cDNA sample) to determine amplification efficiency for each primer set.
Step 4: Data Analysis
- Calculate qPCR ∆Ct values: Ct(target gene) - Ct(reference housekeeping gene). For higher expression, ∆Ct is lower (more negative).
- Normalize RNA-seq counts from the matrix. Use Transcripts Per Million (TPM) or counts normalized by a stable housekeeping gene.
- Perform correlation analysis. Plot log2(TPM + 1) vs. -∆Ct for each gene across samples.
- Calculate Pearson and/or Spearman correlation coefficients.
Representative Quantitative Data
Table 1: Example qPCR Correlation Results for RNA-seq Validation
| Sample Set (n) | # Genes Tested | Pearson Correlation (r) | Spearman Rank Correlation (ρ) | Normalization Method Used |
|---|---|---|---|---|
| HeLa Cells (3) | 30 | 0.92 ± 0.03 | 0.89 ± 0.04 | TPM |
| Mouse Liver (5) | 25 | 0.87 ± 0.05 | 0.91 ± 0.03 | DESeq2 Median-of-Ratios |
| Cancer Cell Panel (6) | 50 | 0.94 ± 0.02 | 0.93 ± 0.02 | TPM |
Title: qPCR Validation Workflow for Gene Count Matrix
Spike-in Control Validation
Core Principle
Commercially available synthetic RNA molecules (spike-ins) are added in known, staggered concentrations to the total RNA sample prior to library preparation. The accuracy of the bioinformatic pipeline is assessed by how linearly the resulting read counts from the gene count matrix reflect the known input abundances of these spike-ins.
Detailed Protocol
Step 1: Spike-in Kit Selection & Addition
- Select a validated spike-in kit (e.g., ERCC ExFold RNA Spike-In Mixes, SIRVs, Sequins).
- These mixes contain 50-100+ synthetic transcripts with defined log-fold concentration differences.
- Critical: Add a constant, small volume (e.g., 1-2 µL) of the diluted spike-in mix to a fixed amount (e.g., 1 µg) of your total RNA sample before any cDNA synthesis or library preparation steps. Mix thoroughly.
Step 2: Library Preparation & Sequencing
- Proceed with your standard RNA-seq library protocol (poly-A selection, rRNA depletion, etc.).
- Note: The spike-in sequences are designed to be compatible with eukaryotic library prep.
- Pool and sequence libraries as usual. Ensure sufficient sequencing depth to detect low-abundance spike-ins.
Step 3: Bioinformatics Processing
- Alignment: Include the spike-in sequence files (FASTA) as an additional reference during genome alignment (e.g., with STAR or HISAT2) or handle as a separate alignment.
- Counting: Generate counts for spike-in sequences alongside endogenous genes using your counting tool (e.g., featureCounts, HTSeq-count).
- Matrix Generation: Your gene count matrix should include a dedicated section for spike-in transcript counts.
Step 4: Data Analysis & Assessment
- Plot observed log2(counts + 1) vs. expected log2(known input concentration) for all spike-in transcripts.
- Perform linear regression. The R² value and slope indicate accuracy and dynamic range.
- Assess sensitivity: determine the lowest input concentration spike-in that is reliably detected (count > 0).
- Assess fold-change accuracy: compare observed vs. expected ratios between spike-in pairs.
Representative Quantitative Data
Table 2: Example Spike-in Control Performance Metrics
| Spike-in Mix | # Spikes | Linear Regression R² (Mean) | Dynamic Range (Log10) | Sensitivity (Lowest Attomole Detected) |
|---|---|---|---|---|
| ERCC ExFold (1:100) | 92 | 0.98 ± 0.01 | 10⁶ | 0.1 amol |
| SIRV Set 3 (E0) | 69 | 0.99 ± 0.005 | 10⁷ | 0.01 amol |
| Sequins (RNA Mix A) | 38 | 0.97 ± 0.02 | 10⁵ | 1 amol |
Title: Spike-in Control Validation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Validation Experiments
| Item / Reagent | Function in Validation | Example Product(s) |
|---|---|---|
| qPCR-Grade Reverse Transcriptase | Converts RNA to cDNA with high fidelity and efficiency for accurate qPCR templates. | SuperScript IV, PrimeScript RT. |
| Validated qPCR Assay Mix | Provides fluorescence detection of amplified DNA (intercalating dye or probe-based). | Power SYBR Green, TaqMan Gene Expression Master Mix. |
| Validated qPCR Primers/Probes | Gene-specific oligonucleotides for targeted amplification and detection. | IDT PrimeTime qPCR Assays, Thermo Fisher TaqMan Assays. |
| Synthetic RNA Spike-in Mix | Provides exogenous RNA standards at known concentrations for absolute calibration. | ERCC ExFold RNA Spike-In Mixes (Thermo Fisher), SIRVs (Lexogen), Sequins. |
| Nucleic Acid Quantitation Kit | Precisely measures RNA and cDNA concentration (critical for normalization). | Qubit RNA BR Assay, Quant-iT RiboGreen. |
| High-Sensitivity DNA Analysis Kit | Assesses library fragment size and quality before sequencing. | Agilent High Sensitivity DNA Kit (Bioanalyzer/TapeStation). |
Thesis Context: This document provides detailed application notes and protocols for the generation of a gene count matrix from BAM files, a critical step in RNA-Seq analysis for transcriptomic studies in biomedical research and drug development.
A gene count matrix, where rows represent genes and columns represent samples, is the fundamental input for differential gene expression analysis. The process of generating this matrix from aligned sequencing reads (BAM files) involves assigning each read to a genomic feature (e.g., a gene), a step known as read quantification. Three predominant methodological approaches exist: direct alignment-based counting (featureCounts, HTSeq) and pseudoalignment-based quantification (Kallisto, Salmon).
The following table summarizes the key characteristics, performance metrics, and recommended use cases for each tool category based on current benchmarking studies.
Table 1: Comparative Summary of Quantification Tools
| Aspect | featureCounts (Subread) | HTSeq | Pseudoalignment (Kallisto/Salmon) |
|---|---|---|---|
| Core Method | Alignment-based; counts reads overlapping exonic regions. | Alignment-based; parses alignments based on strict positional overlap. | Alignment-free; uses k-mer matching to transcriptomes. |
| Input | SAM/BAM files + GTF/GFF annotation. | SAM/BAM files + GTF/GFF annotation. | Raw FASTQ files or SAM/BAM; transcriptome index. |
| Speed | Very Fast (~5-10x faster than HTSeq). | Slow. | Extremely Fast (~20-30x faster than alignment-based). |
| Memory Use | Moderate. | Low-Moderate. | Low. |
| Accuracy | High, standard for genomic alignment-based counts. | High, but can be conservative, discarding ambiguous reads. | High, often superior in transcript-level estimates; effective for isoform-level analysis. |
| Multi-mapping Reads | Can be probabilistically distributed (via --fracOverlap). |
Default mode discards ambiguous reads (mode=union). |
Built-in probabilistic resolution using EM algorithm. |
| Strandedness | Full support for stranded protocols. | Full support for stranded protocols. | Full support for stranded protocols. |
| Dependency | Requires pre-aligned BAM files. | Requires pre-aligned BAM files. | Can run independently of genomic alignment. |
| Best For | Gene-level counts from genomic alignments; standard bulk RNA-Seq pipelines. | Simple, precise gene-level counting where ambiguity is undesirable. | Rapid analysis, large datasets, transcript-level quantification, re-analysis of public data. |
Table 2: Typical Resource Usage (Human RNA-Seq, 30M reads/sample)
| Tool | CPU Time | RAM (GB) | Output Level |
|---|---|---|---|
| featureCounts | 5-10 minutes | 4-8 | Gene |
| HTSeq-count | 45-60 minutes | 2-4 | Gene |
| Salmon (align-mode) | 8-12 minutes | 4-6 | Transcript |
Experimental Protocols
Protocol 1: Gene Counting with featureCounts
Objective: Generate a gene count matrix from a set of BAM files using featureCounts.
Materials: See "Scientist's Toolkit" section. Software: Subread package (featureCounts v2.0+), SAMtools.
Procedure:
- Input Preparation: Ensure all BAM files are sorted and indexed (e.g.,
samtools sort -o sample.sorted.bam sample.bam; samtools index sample.sorted.bam). - Annotation File: Obtain a species-appropriate GTF file (e.g., from GENCODE or Ensembl).
- Run featureCounts: Execute a command to process multiple files simultaneously.
- Output Processing: The primary output
gene_counts.txtcontains a count matrix. The auxiliary filegene_counts.txt.summarycontains assignment statistics. Use the count matrix file (columns 7 onward) for downstream analysis in R/Bioconductor (e.g., DESeq2, edgeR).
Protocol 2: Gene Counting with HTSeq-count
Objective: Generate gene counts using the htseq-count script.
Materials: See "Scientist's Toolkit". Software: HTSeq (v0.13.5+), Python 3, SAMtools.
Procedure:
- Input Preparation: As in Protocol 1, use sorted BAM files. HTSeq can also read SAM files. For BAM, use
-f bamflag. - Run HTSeq-count: Process samples individually, then combine.
- Batch Processing & Matrix Creation: Use a shell loop or workflow system to process all samples. Combine individual count files using a script (e.g., in R or Python) to create the final count matrix, removing the summary lines at the end of each file.
Protocol 3: Quantification with Salmon (Pseudoalignment)
Objective: Perform transcript-level quantification directly from FASTQ files using Salmon, then aggregate to gene-level.
Materials: See "Scientist's Toolkit". Software: Salmon (v1.9+).
Procedure:
- Indexing: Build a decoy-aware transcriptome index (best practice).
- Quantification: Run Salmon in mapping-based mode if you have BAM files, or direct from FASTQ.
- Creating a Gene Matrix: Salmon outputs transcript-level counts (
quant.sf). Use thetximportR/Bioconductor package to import these estimates, summarize them to the gene level, and correct for potential length bias, producing a gene count matrix suitable for DESeq2 or edgeR.
Visualizations
Title: RNA-Seq Quantification Workflow Comparison
Title: Read Assignment Logic for Ambiguous Reads
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions & Materials
| Item | Function/Description |
|---|---|
| High-Quality Reference Genome & Annotation (GTF/GFF) | Essential for alignment and feature identification. Source: ENSEMBL, GENCODE, or UCSC. Must match genome build used in alignment. |
| Alignment Software (STAR, HISAT2) | Generates BAM files from FASTQ for input to featureCounts/HTSeq. STAR is the current standard for spliced alignment. |
| Subread Package (featureCounts) | Provides the featureCounts executable. A fast, efficient counting program. |
| HTSeq Python Framework | Provides the htseq-count script. Offers fine-grained control over counting logic. |
| Salmon / Kallisto | Pseudoalignment tools for ultra-fast transcript-level quantification from FASTQ. |
| SAMtools | Utilities for manipulating SAM/BAM files (sorting, indexing, viewing). Required for preparing BAM inputs. |
| tximport R/Bioconductor Package | Critical for importing transcript-level abundances (from Salmon/Kallisto) and summarizing to gene-level counts for downstream analysis. |
| DESeq2 / edgeR R/Bioconductor Packages | Standard tools for differential expression analysis using the generated gene count matrix. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | RNA-Seq data processing is computationally intensive. Multi-core CPUs and sufficient RAM (≥32GB) are recommended. |
1. Introduction Within the broader thesis on creating a gene count matrix from BAM files, a critical and often overlooked step is the choice of counting methodology. This Application Note details how the selection of a counting tool and strategy (e.g., exon-based, transcript-based, multi-mapping handling) directly impacts the numerical composition of the gene count matrix, thereby influencing the statistical power, false discovery rate, and biological conclusions of subsequent differential expression (DE) analysis.
2. Comparative Quantitative Data: Tool Performance Metrics Table 1: Comparison of Read Counting Tools on a Simulated RNA-seq Dataset (Human, 100M paired-end reads). Data sourced from recent benchmark studies (2023-2024).
| Tool | Counting Paradigm | Time (min) | Memory (GB) | Recall (%) | Precision (%) | Key Differentiating Feature |
|---|---|---|---|---|---|---|
| featureCounts | Gene-Exon | 22 | 4.5 | 95.1 | 97.8 | Fast, directly assigns to genomic features. |
| HTSeq | Gene-Exon | 58 | 6.1 | 94.8 | 98.0 | Conservative, strict intersection-nonempty mode. |
| Salmon | Transcript | 18 (map+quant) | 5.2 | 96.5 | 96.0 | Pseudo-alignment, fast, models abundance. |
| Kallisto | Transcript | 12 (pseudoalign+quant) | 4.8 | 96.2 | 96.3 | Pseudo-alignment, ultra-fast bootstrap. |
| RSEM | Transcript | 95 | 7.5 | 96.8 | 96.5 | Detailed model, estimates isoform expression. |
Table 2: Downstream DE Impact of Counting Strategies on Overlapping Genes (Simulated Data, n=3 vs. 3).
| Counting Strategy / Tool | Total DE Genes (FDR<0.05) | False Positives | False Negatives | Notable Effect on Specific Gene Class |
|---|---|---|---|---|
| featureCounts (default) | 1250 | 89 | 102 | Under-counts overlapping genes. |
| featureCounts (--countReadPairs) | 1275 | 85 | 105 | Better for paired-end, slight increase in counts. |
| Salmon (--gencode) | 1315 | 78 | 75 | Higher sensitivity for multi-isoform genes. |
| HTSeq (intersection-strict) | 1190 | 65 | 125 | Most conservative, highest false negatives. |
| Kallisto (--bias) | 1305 | 82 | 80 | Corrects sequence bias, affects GC-rich genes. |
3. Experimental Protocols
Protocol 1: Generating a Gene Count Matrix with featureCounts (Exon-Based) Objective: To generate a raw gene-level count matrix from aligned BAM files. Materials: High-performance computing cluster, BAM files, reference genome annotation (GTF/GFF). Procedure:
- Software Installation: Install Subread package v2.0.7 or later via conda:
conda install -c bioconda subread. - Input Preparation: Ensure BAM files are sorted and indexed. Prepare a text file (
bam_list.txt) with full paths to all BAM files. - Run featureCounts: Execute the following command:
featureCounts -T 8 -p --countReadPairs -a Homo_sapiens.GRCh38.109.gtf -o gene_counts.txt -g gene_id -s 1 bam_list.txt-T 8: Use 8 CPU threads.-p: Count fragments (for paired-end data).--countReadPairs: Count read pairs instead of reads.-s 1: Strand-specific counting (forward strand).
- Matrix Extraction: The primary output
gene_counts.txtcontains the count matrix. Use thecutcommand to extract only the columns with counts for downstream analysis.
Protocol 2: Generating a Gene Count Matrix via Transcript-Abundance Tools (Salmon) Objective: To generate a gene-level count matrix via transcript quantification, enabling correction for sequence bias and GC content. Materials: As in Protocol 1, plus a transcriptome decoy-aware index. Procedure:
- Index Building: Download cDNA and genome FASTA files. Build a selective alignment index:
salmon index -t transcriptome.fa -d decoys.txt -i salmon_index -p 12. - Quantification: Run Salmon in mapping-based mode for each sample:
salmon quant -i salmon_index -l A -1 sample_1.fastq.gz -2 sample_2.fastq.gz -p 8 --gcBias --seqBias -o quants/sample_A--gcBias/--seqBias: Model and correct for respective biases.
- Aggregation to Gene Level: Use
tximportin R to summarize transcript abundances to gene-level counts, accounting for length and abundance biases.
4. Signaling Pathways & Workflow Visualizations
Diagram Title: Counting Choice Influences DE Analysis Pathway
Diagram Title: Gene Count Matrix Creation Decision Workflow
5. The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Solution | Function / Purpose in Experiment |
|---|---|
| Subread (featureCounts) | A highly efficient and fast read summarization program that assigns sequencing reads to genomic features (e.g., genes, exons). Core tool for direct exon-based counting. |
| Salmon | A bias-aware transcript-level quantification tool that uses a dual-phase mapping and quantification model. Corrects for technical biases to improve accuracy. |
| tximport R/Bioconductor Package | Critical for bridging transcript-level quantifications (from Salmon/Kallisto) to gene-level DE analysis. Corrects for changes in gene length across samples, enabling use of count-based statistical models. |
| GENCODE Comprehensive Annotation | A high-quality, comprehensive reference gene annotation set. Provides the definitive gene and transcript models required for accurate read assignment. More reliable than older RefSeq entries. |
| DESeq2 R/Bioconductor Package | The standard DE analysis tool that models raw counts using a negative binomial distribution. The choice of count matrix directly affects its dispersion estimation and statistical testing. |
| Decoy Sequence File | Used during Salmon indexing to "capture" reads that map outside annotated transcripts, reducing spurious mapping and improving quantification accuracy of expressed transcripts. |
| Strand-Specific Library Prep Kits | (e.g., Illumina Stranded mRNA Prep). The library preparation chemistry determines the -s parameter in counting tools, which is critical for accurate assignment and avoiding anti-sense inflation. |
Within the broader thesis research on How to create a gene count matrix from BAM files, a critical step is the selection of an appropriate quantification tool. The process of generating a gene count matrix involves aligning sequencing reads to a reference genome (resulting in BAM files) and then assigning these aligned reads to genomic features (e.g., genes). Multiple software tools exist for this quantification step, each employing different algorithms (e.g., overlap-based, intersection-based). This application note provides a framework for benchmarking these tools based on three pillars—Speed, Accuracy, and Reproducibility—to inform robust, scalable, and reliable downstream differential expression analysis in drug discovery and basic research.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item/Category | Example(s) | Function in Gene Count Matrix Generation |
|---|---|---|
| Quantification Software | featureCounts, HTSeq-count, Salmon (alignment-based mode), RSEM, kallisto (pseudoalignment) | Core tool that reads BAM/SAM files and a GTF annotation to assign reads to genes, generating raw counts. |
| Reference Genome & Annotation | GENCODE, Ensembl, RefSeq | Provides the coordinate system (FASTA) and gene model definitions (GTF/GFF) essential for accurate read assignment. |
| Alignment File | Coordinate-sorted BAM file with index (.bai) | The input file containing reads mapped to the reference genome. Must be sorted for most tools. |
| Benchmarking Dataset | SEQC/MAQC-III, Simulated RNA-seq data from Polyester | Gold-standard or synthetic data with known expression truth for accuracy assessment. |
| Containerization Tool | Docker, Singularity/Apptainer | Ensures reproducibility by encapsulating the exact software environment, including all dependencies. |
| Workflow Management System | Nextflow, Snakemake | Orchestrates the multi-step benchmarking pipeline, managing software calls and data flow. |
| High-Performance Computing (HPC) | SLURM cluster, Cloud computing (AWS, GCP) | Provides the computational resources necessary for large-scale benchmarking of speed and scalability. |
Experimental Protocols for Benchmarking
Protocol 3.1: Benchmarking Setup and Data Preparation
- Select Benchmarking Tools: Choose a set of candidate quantification tools (e.g., featureCounts, HTSeq, RSEM).
- Obtain Ground Truth Data: Download a validated reference dataset (e.g., SEQC project's sample A vs B) or generate a synthetic dataset using a simulator like Polyester with a known count matrix.
- Prepare Input Files: Ensure all BAM files are coordinate-sorted and indexed. Use a consistent, high-quality GTF annotation file (e.g., from GENCODE) for all tools.
- Containerize Tools: Create Docker/Singularity images for each tool, specifying exact software versions and dependencies. Record all images with immutable tags.
Protocol 3.2: Measuring Speed and Computational Efficiency
- Execution: Run each tool on an identical HPC node (specifying CPU/memory constraints) using the same set of BAM files.
- Timekeeping: Use the
/usr/bin/time -vcommand to record elapsed wall-clock time, peak memory usage (RSS), and CPU utilization for each run. - Replication: Repeat timing measurements across multiple BAM files of varying sizes (e.g., 10M, 50M, 100M reads) and in triplicate to account for system noise.
- Data Collection: Log all metrics for comparative analysis.
Protocol 3.3: Assessing Accuracy
- Run Quantification: Execute each containerized tool on the benchmarking dataset (with known truth) to generate predicted count matrices.
- Normalization: For comparison, normalize all predicted and truth matrices to counts per million (CPM) or using a variance-stabilizing transformation.
- Calculate Metrics: Compare the predicted counts to the known truth using:
- Correlation: Pearson and Spearman correlation coefficients of gene-level counts.
- Error Metrics: Mean Absolute Error (MAE) or Root Mean Square Error (RMSE) on log2-transformed normalized counts.
- Differential Expression (DE) Recovery: Perform DE analysis (e.g., using DESeq2) on the predicted counts and compare the list of significant genes to the truth set using precision, recall, and F1-score.
Protocol 3.4: Evaluating Reproducibility
- Environment Reproducibility: Re-run the entire benchmarking pipeline on a different computing system using only the provided container images and workflow scripts.
- Technical Replicate Consistency: Process the same BAM file multiple times (n=5) with each tool in an identical environment. Calculate the coefficient of variation (CV) for expression counts of high- and medium-abundance genes.
- Documentation: Record all parameters, software versions, and random seeds in a structured metadata file (e.g., JSON, YAML) alongside output files.
Table 1: Benchmarking Results for Selected Quantification Tools (Hypothetical Data Based on Current Tool Profiles).
| Tool | Avg. Speed (min per 10M reads) | Peak Memory (GB) | Correlation with Truth (Pearson r) | DE F1-Score | Count CV across Replicates |
|---|---|---|---|---|---|
| featureCounts | 2.1 | 1.8 | 0.988 | 0.96 | 0.001 |
| HTSeq | 15.7 | 4.5 | 0.985 | 0.95 | 0.001 |
| RSEM | 22.3 | 6.2 | 0.992 | 0.97 | 0.005 |
| Salmon (align) | 8.5 | 3.1 | 0.990 | 0.96 | 0.002 |
Note: Data is illustrative. Actual results depend on dataset, genome, and parameters. Speed/memory tested on a single 16-core node.
Visualized Workflows and Relationships
Title: Gene Count Tool Benchmarking Workflow
Title: Tool Selection Logic Based on Benchmarking Goals
Best Practices for Reproducible and FAIR-Compliant Count Matrix Generation
Within a thesis investigating how to create a gene count matrix from BAM files, the step of quantification is critical. This application note details a protocol for generating a gene-level count matrix from aligned sequencing reads (BAM files) that adheres to FAIR (Findable, Accessible, Interoperable, Reusable) principles and ensures full computational reproducibility.
Table 1: Comparison of High-Performance Read Quantification Tools
| Tool | Primary Method | Reference Handling | Output Compatibility | Key Strength for FAIR |
|---|---|---|---|---|
| HTSeq | Overlap of aligned reads with genomic features. | Requires sorted GTF/GFF. | Simple count table. | Conceptual clarity; well-established protocol. |
| featureCounts | Ultra-fast overlap using read summarization program. | Directly uses GTF/GFF. | Count matrix, statistical summaries. | Speed and integrated QC metrics. |
| RSEM | Expectation-Maximization for isoform-resolution. | Can use transcriptome FASTA/GTF. | Expected counts, TPM, FPKM. | Models uncertainty in read assignment. |
| Salmon / kallisto | Pseudo-alignment / lightweight mapping. | Requires decoy-aware transcriptome index. | Estimated counts, TPM. | Extreme speed and accuracy for transcript-level analysis. |
Detailed Protocol: featureCounts-Based Quantification
Objective: To generate a gene-level count matrix from stranded, paired-end RNA-seq BAM files. FAIR Alignment: This protocol uses versioned software, explicit parameters, and generates standard output formats for Interoperability and Reusability.
Materials & Inputs:
- Sorted BAM Files: One per sample, coordinate-sorted, with index files (.bai).
- Genome Annotation File: A versioned Gene Transfer Format (.gtf) file from a reference database (e.g., Ensembl, GENCODE).
- Sample Metadata Sheet: A CSV file detailing experimental conditions, library preparation details, and BAM file paths.
Procedure:
- Environment Setup: Create a Conda environment with the required tool.
- Execute featureCounts: Run in a single command for all samples to ensure consistent parameters.
- Generate Summary Metadata: Capture the exact command and environment.
- Post-processing: Use a scripting language (e.g., R, Python) to extract the count matrix, merging it with sample metadata.
Workflow Visualization
Title: FAIR Count Matrix Generation Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents & Materials for Quantification
| Item | Function & Relevance to FAIR/Reproducibility |
|---|---|
| Reference Genome Assembly | Provides the coordinate system for alignment (e.g., GRCh38.p14). Must be versioned. |
| Gene Annotation File (GTF/GFF3) | Defines genomic features (genes, exons). Source and version are critical metadata. |
| Conda/Bioconda Channel | Provides version-controlled, reproducible software environments for tools like Subread/featureCounts. |
| Unique Molecular Identifiers (UMIs) | If present in the data, require specific handling in quantification to correct for PCR duplicates. |
| Sample Metadata Schema | A structured template (e.g., following MIAME/MINSEQE) to capture sample, protocol, and data relationships. |
| Compute Environment Snapshot | Tools like Docker/Singularity or Conda export files that encapsulate the exact software stack used. |
Conclusion
Creating an accurate and reliable gene count matrix from BAM files is a foundational, yet complex, step that directly influences all subsequent genomic analyses. By mastering the foundational concepts, implementing a robust methodological workflow, proactively troubleshooting common issues, and rigorously validating outputs, researchers can ensure the integrity of their data for critical applications in differential expression, biomarker discovery, and therapeutic target identification. As single-cell and long-read sequencing evolve, the principles of careful quantification remain paramount, underscoring the need for standardized, optimized pipelines to drive reproducible discoveries in biomedical and clinical research.