STAR RNA-seq Alignment: A Complete Guide for Researchers from Basics to Advanced Analysis
This comprehensive guide provides researchers, scientists, and drug development professionals with a complete workflow for performing RNA-seq read alignment using the STAR (Spliced Transcripts Alignment to a Reference) aligner.
STAR RNA-seq Alignment: A Complete Guide for Researchers from Basics to Advanced Analysis
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with a complete workflow for performing RNA-seq read alignment using the STAR (Spliced Transcripts Alignment to a Reference) aligner. We cover foundational concepts of alignment theory and STAR's algorithm, present a detailed step-by-step protocol from installation to execution, address common troubleshooting scenarios and optimization strategies for computational efficiency, and explore validation methods and comparisons with other aligners. The article equips readers with both practical skills and critical knowledge to ensure accurate, reproducible results for downstream differential expression, isoform discovery, and clinical biomarker identification.
RNA-seq Alignment Fundamentals: Understanding STAR's Algorithm and Prerequisites
RNA sequencing (RNA-seq) is a high-throughput technology that provides a comprehensive snapshot of the transcriptome. Accurate read alignment is the foundational computational step that maps sequencing reads to a reference genome, directly impacting all downstream analyses, including differential gene expression, isoform discovery, and variant calling. Misalignment can introduce severe biases, leading to incorrect biological interpretations. This application note details protocols for performing robust read alignment using the Spliced Transcripts Alignment to a Reference (STAR) aligner within a thesis research framework.
The Criticality of Alignment Accuracy: A Quantitative Perspective
The performance of an aligner is measured by its speed, memory usage, and, most critically, its accuracy in placing reads, especially across splice junctions. The following table summarizes key metrics from recent benchmark studies comparing leading aligners.
Table 1: Comparative Performance of RNA-seq Aligners (Simulated Human Data)
| Aligner | Alignment Speed (M reads/hour) | Memory Usage (GB) | Sensitivity (%) | Precision (%) | Splice Junction Recall (%) |
|---|---|---|---|---|---|
| STAR | 110 | 28 | 96.5 | 95.8 | 94.2 |
| HISAT2 | 130 | 8.5 | 95.1 | 96.2 | 92.7 |
| TopHat2 | 20 | 4 | 90.3 | 97.1 | 88.5 |
| Salmon (alignment-free) | >500 | 5 | - | - | - |
Data synthesized from current benchmarking literature (2023-2024). Sensitivity/Precision refer to overall read placement. STAR demonstrates an optimal balance of high sensitivity and splice junction detection, which is paramount for novel isoform analysis.
Protocol: Read Alignment with STAR for RNA-seq Research
This protocol is designed for thesis-level research, emphasizing reproducibility and accuracy for mammalian genomes.
Part 1: Generating the Genome Index
A one-time indexing of the reference genome and annotation is required.
Materials & Reagents:
- Reference Genome FASTA: Primary assembly file (e.g.,
GRCh38.primary_assembly.fa). - Gene Annotation GTF: Comprehensive annotation file (e.g.,
gencode.v44.annotation.gtf). - Computing Resources: Server with ≥ 32 GB RAM and multiple CPU cores.
- STAR Software: Version 2.7.11a or later.
Procedure:
- Download and decompress the genome FASTA and annotation GTF files.
- Create a dedicated directory for the genome index:
mkdir STAR_Genome_Index. - Execute the STAR indexing command:
--runThreadN: Number of CPU threads.--sjdbOverhang: Read length minus 1. For 100bp paired-end reads, use 99.
- The process will create numerous files in the
STAR_Genome_Indexfolder. Verify theLog.outfile for successful completion.
Part 2: Performing the Alignment
This step maps the sequencing reads (in FASTQ format) to the indexed genome.
Materials & Reagents:
- RNA-seq Reads: Compressed FASTQ files (e.g.,
SampleA_R1.fastq.gz,SampleA_R2.fastq.gz). - Genome Index: The
STAR_Genome_Indexdirectory from Part 1. - Computing Resources: High-memory node (≥ 32 GB RAM recommended).
Procedure:
- Create an output directory for alignment results:
mkdir -p STAR_Alignments/SampleA. - Execute the STAR alignment command:
--readFilesCommand zcat: For reading gzipped files.--outSAMtype BAM SortedByCoordinate: Outputs a sorted BAM file ready for downstream tools.--quantMode GeneCounts: Outputs read counts per gene intoReadsPerGene.out.tab.--twopassMode Basic: Enables more sensitive novel junction discovery, recommended for thesis research.
- Key output files include:
Aligned.sortedByCoord.out.bam: The primary alignment file.Log.final.out: Summary alignment statistics (critical for QA).ReadsPerGene.out.tab: Raw count matrix input for differential expression.
Part 3: Post-Alignment Quality Assessment
Protocol: Use MultiQC to aggregate STAR logs and other QC tool outputs.
Review the report for metrics like % of reads uniquely mapped and % of reads mapped to multiple loci.
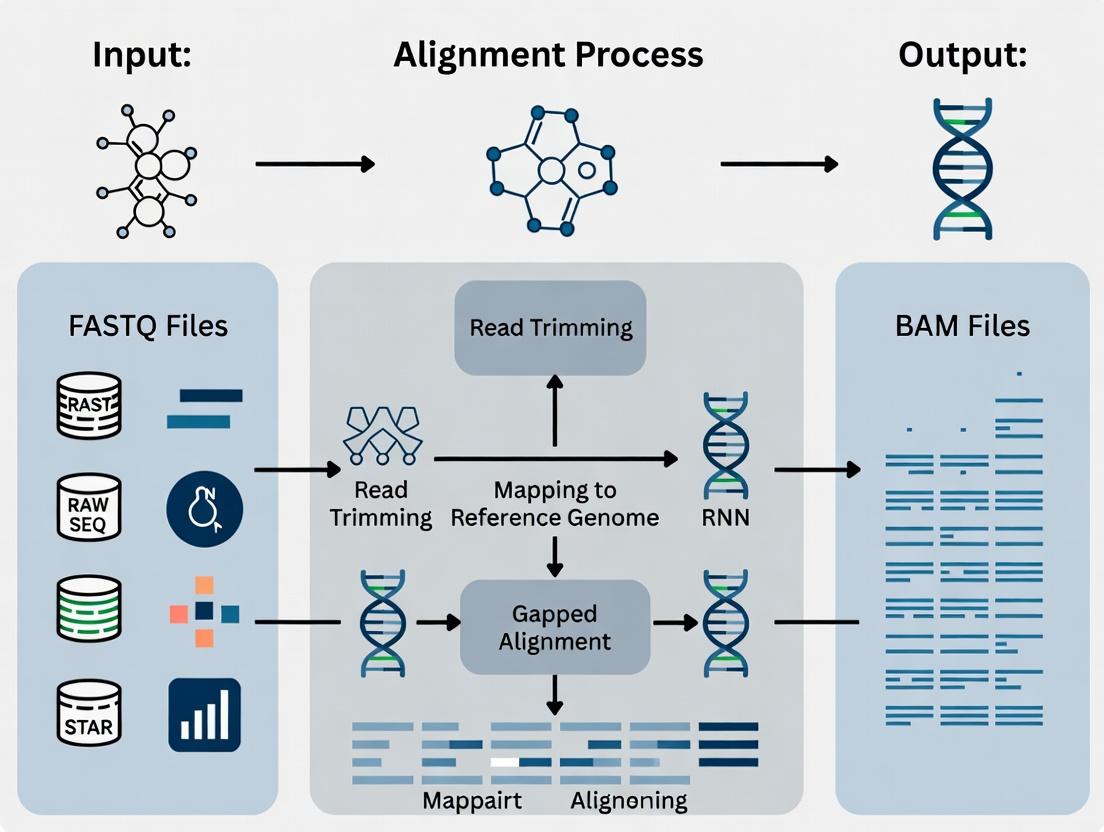
Visualizing the RNA-seq Workflow and STAR's Role
RNA-seq Analysis Pipeline with STAR
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for RNA-seq Alignment Research
| Item | Function & Rationale |
|---|---|
| High-Quality Total RNA (RIN > 8) | Intact RNA minimizes bias in library preparation, ensuring even coverage across transcripts. |
| Strand-Specific Library Prep Kit (e.g., Illumina TruSeq Stranded) | Preserves the orientation of transcripts, allowing determination of the originating DNA strand, crucial for antisense and overlapping gene analysis. |
| STAR Aligner Software | Specialized for splice-aware alignment, offering high accuracy and speed for large genomes. Critical for thesis work involving novel junction detection. |
| Reference Genome & Annotation (e.g., GENCODE, RefSeq) | The mapping blueprint. Using the primary assembly and comprehensive annotation (GTF) is essential for accurate placement and quantification. |
| SAMtools/BEDTools | Essential software suites for processing, indexing, and interrogating alignment (BAM) files post-STAR alignment. |
| High-Performance Computing (HPC) Node | Alignment is computationally intensive. Access to servers with significant RAM (≥ 32 GB) and multi-core CPUs is mandatory for timely thesis research. |
Why STAR? Advantages for Spliced Alignment and Handling Junctions
In the context of RNA-seq read alignment, the STAR (Spliced Transcripts Alignment to a Reference) aligner represents a critical methodological advancement. Unlike traditional DNA-seq aligners, STAR is explicitly engineered to handle the non-contiguous nature of RNA-seq reads caused by splicing. Its core algorithm uses a sequential two-step process: first, seed searching based on a compressed suffix array, followed by precise stitching and scoring of clusters of seed matches to identify spliced alignments across exon junctions. This design confers significant advantages in speed, sensitivity, and accuracy for junction discovery, making it the de facto standard for modern transcriptomic studies.
Advantages of STAR for Spliced Alignment
Unmatched Mapping Speed
STAR utilizes an uncompressed suffix array index for the reference genome, enabling extremely rapid exact matching of read segments. Its algorithmic efficiency allows it to process ~30 million paired-end reads per hour on a standard 12-core server, drastically reducing computational time compared to earlier splice-aware aligners.
High Sensitivity for Novel Junctions
STAR can detect canonical, non-canonical, and novel splice junctions without prior annotation. It does this by clustering and stitching matching sequences (Maximal Mappable Prefixes) from different genomic locations. This sensitivity is crucial for discovering novel isoforms, fusion genes, and mutations in splicing patterns in disease research.
Precision in Junction Boundary Determination
The aligner employs precise alignment and scoring for the junctions themselves, reporting the exact intronic coordinates. This precision is vital for downstream analyses like differential isoform usage and mutation detection near splice sites.
Integrated Output for Downstream Analysis
STAR outputs alignment files (BAM), unmapped reads, and junction tables in a single run. Its junction output includes read counts per junction and intron motifs, providing immediate data for splice event quantification.
Table 1: Quantitative Performance Comparison of RNA-seq Aligners
| Aligner | Algorithm Type | Relative Speed | Novel Junction Detection | Memory Footprint (Human Genome) | Key Outputs |
|---|---|---|---|---|---|
| STAR | Seed-and-extend (Suffix Array) | ~30-50M reads/hr* | Yes | High (~32 GB) | BAM, Junctions, Transcriptome BAM |
| HISAT2 | Hierarchical Graph FM-index | ~15-25M reads/hr | Yes | Moderate (~12 GB) | BAM, Splice sites |
| TopHat2 | Bowtie2-based + segmentation | ~5-10M reads/hr | Limited | Low (~4 GB) | BAM, Junctions |
| BBMap | k-mer based | ~20-40M reads/hr | Limited | High (~25 GB) | BAM |
*Performance on a 12-core CPU system. Data compiled from recent benchmarking studies.
Detailed Protocol: RNA-seq Read Alignment with STAR
Part A: Generating the Genome Index
- Objective: Create a reference genome index optimized for STAR's suffix array search.
- Inputs: Reference genome FASTA file(s); annotation file (GTF/GFF) – optional but recommended.
- Protocol:
- Gather Files: Download reference genome (e.g., GRCh38.p14 from GENCODE) and corresponding annotation GTF.
- Execute Indexing Command:
- --sjdbOverhang: Critical parameter. Should be (Read Length - 1). For 100bp reads, use
99. - --runThreadN: Number of CPU threads to use.
Part B: Performing the Alignment
- Objective: Map RNA-seq reads (FASTQ) to the indexed genome, outputting alignments and junction information.
- Inputs: Paired-end or single-end FASTQ files; STAR genome index.
- Protocol:
- Navigate: Go to the directory containing your FASTQ files.
- Execute Alignment Command (Paired-end example):
- --quantMode TranscriptomeSAM: Outputs alignments translated into transcript coordinates for RSEM/StringTie.
- --quantMode GeneCounts: Counts reads per gene directly from the alignment.
- --outFilterMultimapNmax: Maximum number of loci a read can map to. Higher values increase sensitivity but reduce precision.
Part C: Output Interpretation and Junction Analysis
- Key Output Files:
*Aligned.sortedByCoord.out.bam: Sorted alignment file for visualization and downstream analysis.*ReadsPerGene.out.tab: Raw gene-level read counts.*SJ.out.tab: Junction file – the critical output for splice analysis.
- Junction File (
SJ.out.tab) Columns:- Chromosome
- Intron start (1-based)
- Intron end (1-based)
- Strand (0: undefined, 1: +, 2: -)
- Intron motif (e.g., GT/AG)
- Annotated (0: unannotated, 1: annotated)
- Unique mapping read count
- Multi-mapping read count
- Maximum splice alignment overhang
Visualizing the STAR Alignment Workflow and Logic
STAR RNA-seq Alignment and Analysis Workflow
The Scientist's Toolkit: Key Research Reagent Solutions for RNA-seq with STAR
Table 2: Essential Materials and Reagents
| Item | Function in RNA-seq/STAR Protocol | Example/Note |
|---|---|---|
| High-Quality Total RNA | Starting material. RIN > 8.0 recommended for mRNA-seq. | Isolated via column-based kits (e.g., Qiagen RNeasy). |
| Poly-A Selection or rRNA Depletion Kits | Enriches for mRNA or removes ribosomal RNA. Crucial for library prep. | NEBNext Poly(A) mRNA Magnetic Kit; Illumina Ribo-Zero. |
| Strand-Specific Library Prep Kit | Preserves strand orientation of transcripts for accurate isoform assignment. | Illumina Stranded mRNA Prep; NEBNext Ultra II Directional. |
| High-Fidelity DNA Polymerase | Amplifies cDNA libraries with minimal bias for accurate quantification. | KAPA HiFi HotStart ReadyMix; Q5 High-Fidelity DNA Polymerase. |
| Size Selection Beads | Purifies and selects cDNA fragments of desired length (e.g., ~300bp insert). | SPRIselect beads (Beckman Coulter). |
| STAR Aligner Software | Core tool for performing spliced alignment of RNA-seq reads. | Downloaded from https://github.com/alexdobin/STAR. |
| High-Performance Computing (HPC) Resources | Required for STAR's genome indexing (~32GB RAM) and fast alignment. | Server with ≥16 cores and ≥64GB RAM recommended. |
| SAMtools/BEDTools | For processing, indexing, and manipulating BAM and junction files post-STAR. | Essential utilities for downstream analysis. |
Application Notes: STAR's Core Algorithm in RNA-Seq Alignment
STAR (Spliced Transcripts Alignment to a Reference) employs a novel strategy for precise, fast alignment of RNA-seq reads. Its core algorithm is designed to address the key challenge of detecting spliced alignments across exon junctions without relying on pre-annotated splice sites.
Algorithmic Stages in the Context of RNA-Seq
The algorithm processes each read through three sequential, interdependent stages. Its efficiency stems from the use of uncompressed suffix arrays derived from the reference genome, enabling rapid seed searching.
Table 1: Quantitative Performance Benchmarks of STAR Algorithm
| Metric | Typical Value | Notes |
|---|---|---|
| Alignment Speed | ~30-45 million reads/hour (30+ CPU threads) | Highly dependent on genome size and compute resources. |
| Memory Footprint | ~30-35 GB for Human (GRCh38) | Primary requirement for loading the genome index. |
| Mapped Read Percentage | 85-95% of total reads | Varies with RNA quality and library prep. |
| Precision of Junction Discovery | >95% for annotated junctions | Based on comparison to reference transcriptomes. |
| De Novo Junction Detection | Capable of identifying novel splice events | Relies on scoring and filtering of clustered segments. |
Key Advantages for Drug Development
For researchers and drug development professionals, STAR's algorithm provides:
- Hypothesis-Free Discovery: Unbiased detection of novel isoforms, fusion transcripts, and genetic variants, crucial for oncology and biomarker research.
- High Sensitivity: The seed-and-vote stage ensures even low-abundance transcripts are captured, important for detecting differentially expressed genes in response to compound treatment.
- Reproducibility: Deterministic alignment logic ensures consistent results across runs, essential for regulatory compliance in preclinical studies.
Experimental Protocols for Key STAR Workflows
Protocol: Generating a Genome Index
This is a prerequisite one-time step for a given reference genome and annotation.
- Input Preparation:
- Gather the reference genome FASTA file(s).
- Obtain the corresponding annotation file in GTF format.
- Command Execution:
--sjdbOverhangshould be set to (read length - 1). A value of 100 is standard for modern long-read sequencing.
- Validation: Check the
Log.outfile for successful completion. The index directory will contain binary files (e.g.,Genome,SAindex).
Protocol: Performing Alignment of RNA-Seq Reads
This is the main alignment protocol using the core algorithm.
- Data Input: Prepare FASTQ files (single- or paired-end).
- Alignment Run:
- Critical Parameters for Algorithm Control:
--seedSearchStartLmax: Controls maximum length of the seed for the seed-and-vote stage (default 50). Lower for higher sensitivity, higher for speed.--seedPerReadNmax: Maximum number of seeds per read (default 10000).--seedPerWindowNmax: Maximum number of seeds per window for clustering (default 50). Adjust to manage memory in dense genomic regions.--outFilterScoreMinOverLread: Minimum alignment score (normalized by read length) for output.--twopassMode Basic: Enables two-pass mapping for novel junction discovery, essential for de novo analysis.
Visualizations of the Core Algorithm
Title: STAR's Three-Stage Alignment Algorithm Workflow
Title: Seed-and-Vote Stage Mechanism
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagent Solutions for STAR RNA-Seq Analysis
| Item | Function in the Workflow | Example/Notes |
|---|---|---|
| High-Quality Total RNA | Starting biological material. Integrity (RIN > 8) is critical for accurate splice variant detection. | Isolated via column-based kits (e.g., Qiagen RNeasy) with DNase I treatment. |
| Strand-Specific Library Prep Kit | Converts RNA to sequencer-compatible cDNA libraries. Preserves strand information for accurate isoform assignment. | Illumina Stranded mRNA Prep, NEBNext Ultra II Directional. |
| STAR Aligner Software | Executes the core alignment algorithm. | Latest version from GitHub (https://github.com/alexdobin/STAR). |
| High-Performance Compute (HPC) Resource | Required for genome indexing and fast alignment due to memory and CPU demands. | ~32+ GB RAM, 10+ CPU cores per run recommended. |
| Reference Genome FASTA | The sequence against which reads are aligned. | Downloaded from ENSEMBL, UCSC, or NCBI (e.g., GRCh38.p13 for human). |
| Gene Annotation File (GTF/GFF3) | Provides known gene models for genome indexing and quantitation. | Matching version to the genome (e.g., GENCODE v44 for GRCh38). |
| Post-Alignment Toolkit | Tools for processing STAR's BAM outputs for downstream analysis. | Samtools for file manipulation, featureCounts/HTSeq for quantification, IGV for visualization. |
| RNA Spike-In Controls | External RNA controls added prior to library prep to monitor technical variability and sensitivity. | ERCC RNA Spike-In Mix (Thermo Fisher). |
Within the thesis "How to perform read alignment with STAR for RNA-seq research," successful execution depends entirely on three foundational prerequisites. This document details the acquisition, validation, and preparation of the reference genome, its structural annotation file (GTF/GFF), and raw sequencing data (FASTQ files) prior to alignment. Proper handling of these components ensures accurate, reproducible, and biologically meaningful RNA-seq analysis.
Reference Genome
The reference genome is a digital nucleotide sequence representing the genetic blueprint of the target organism, serving as the alignment scaffold.
Key Considerations:
- Source & Version: Always obtain genomes from authoritative repositories (e.g., ENSEMBL, NCBI, UCSC). Note the exact assembly version (e.g., GRCh38.p14, GRCm39).
- Integrity: Files should be checked for integrity using MD5 or SHA sums.
- Contaminants: Some genomes include alternative haplotypes or patch sequences; researchers may need to exclude them for standard RNA-seq alignment.
Protocol 1.1: Downloading and Validating a Reference Genome from ENSEMBL
- Navigate to the ENSEMBL FTP site.
- Locate the directory for your organism (e.g.,
release-112/fasta/homo_sapiens/dna/). - Download the primary assembly file, typically named
Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz. - Simultaneously download the corresponding checksum file (
CHECKSUMS). - Validate file integrity using the command:
sum -c CHECKSUMS 2>&1 | grep your_filename.fa.gz. - Uncompress the file:
gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz.
Table 1: Recommended Genomic Data Sources
| Repository | Primary URL | Key Feature | Common File Format |
|---|---|---|---|
| ENSEMBL | ftp.ensembl.org/pub/ | Curated genes & comprehensive annotation | FASTA (.fa, .fasta) |
| NCBI RefSeq | ftp.ncbi.nlm.nih.gov/genomes/ | Non-redundant, reviewed reference sequences | FASTA (.fna) |
| UCSC Genome Browser | hgdownload.soe.ucsc.edu/downloads.html | Mirror of GenBank with UCSC track hubs | FASTA (.fa) |
Genome Annotation File (GTF/GFF)
Annotation files describe the coordinates and metadata of genomic features (genes, transcripts, exons, etc.) in the reference genome. STAR uses this to map splice junctions and quantify gene-level expression.
GTF vs. GFF3: The Gene Transfer Format (GTF) is a specific, widely used variant of the General Feature Format (GFF). For STAR, a comprehensive GTF file is required.
Protocol 1.2: Acquiring a Matching GTF Annotation File
- From the same ENSEMBL release, navigate to the annotation directory (e.g.,
release-112/gtf/homo_sapiens/). - Download the comprehensive GTF file (e.g.,
Homo_sapiens.GRCh38.112.gtf.gz). - Ensure the genome assembly version in the GTF file name matches your downloaded FASTA file exactly.
- Validate and uncompress the file.
Table 2: Critical Attributes in a GTF File for RNA-seq
| Column | Content | Purpose for STAR |
|---|---|---|
| 1: seqname | Chromosome/contig name | Must match FASTA header names. |
| 3: feature | "gene", "exon", "transcript" | Identifies genomic element type. |
| 4,5: start, end | 1-based genomic coordinates | Defines feature location. |
| 7: strand | "+", "-", "." | Indicates transcriptional orientation. |
| 9: attributes | Key-value pairs (e.g., gene_id "ENSG00000139618") |
Essential for gene/transcript grouping. |
Raw FASTQ Files
FASTQ files contain the raw nucleotide sequences (reads) and their corresponding per-base quality scores from the sequencing instrument.
Key Characteristics: Each read is represented by four lines: Sequence ID, nucleotide sequence, a separator (+), and quality scores encoded in Phred+33 (Illumina 1.8+).
Protocol 1.3: Quality Control of Raw FASTQ Files with FastQC
- Install FastQC:
conda install -c bioconda fastqc. - Run a basic quality assessment:
fastqc sample_R1.fastq.gz sample_R2.fastq.gz -o ./fastqc_report/. - Inspect the generated HTML reports. Key metrics to examine:
- Per Base Sequence Quality: Scores should be mostly >30 across all cycles.
- Adapter Content: Percentage of adapter sequence present.
- Sequence Duplication Levels: High duplication may indicate PCR bias.
- Aggregate reports across multiple samples using MultiQC:
multiqc ./fastqc_report/.
Table 3: Essential FASTQ File Metrics Pre-Alignment
| Metric | Optimal Range/Value | Implication for Downstream Analysis |
|---|---|---|
| Read Length | Typically 75-150bp (SE or PE) | Determines alignment speed and specificity. |
| Total Reads | Project-dependent (e.g., 20-40M per sample) | Defines sequencing depth and statistical power. |
| Q20 / Q30 Score | >95% / >90% per sample | Low scores indicate poor base calling. |
| GC Content | Organism-specific (e.g., ~40-50% for human) | Deviations may indicate contamination. |
| Adapter Contamination | < 5% | High levels necessitate more aggressive trimming. |
The Scientist's Toolkit
Table 4: Research Reagent Solutions & Essential Materials
| Item | Function in Pre-Alignment Phase |
|---|---|
| High-Quality Reference Genome (FASTA) | Provides the canonical sequence against which reads are aligned. |
| Curated Annotation File (GTF) | Defines gene models for splice-aware alignment and quantification. |
| Raw RNA-seq Reads (FASTQ) | The primary experimental data containing sequence information. |
| FastQC | Tool for initial quality assessment of FASTQ files. |
| Trimmomatic or Cutadapt | Tools to remove adapter sequences and low-quality bases. |
| SHA/MD5 Checksum | Cryptographic validation ensuring file integrity after transfer. |
| Computational Resources | Adequate CPU, RAM (~32GB+), and storage for genome index generation. |
Visualization: RNA-seq Pre-Alignment Data Preparation Workflow
RNA-seq Pre-Alignment Data Workflow
Meticulous attention to the acquisition, validation, and quality control of the reference genome, annotation file, and raw FASTQ files is a non-negotiable first step. These curated inputs directly determine the accuracy of the subsequent STAR alignment and all downstream analyses in the RNA-seq pipeline. Ensuring version compatibility between genome and annotation, alongside rigorous sequencing data QC, establishes the foundation for robust scientific discovery.
Application Notes
This guide details the hardware and software prerequisites for performing RNA-seq read alignment using the Spliced Transcripts Alignment to a Reference (STAR) aligner, a critical step in transcriptomic analysis for drug discovery and basic research. The recommendations are based on the computational demands of processing high-throughput sequencing data.
Hardware Recommendations
STAR is a memory-intensive application designed for speed. The primary bottleneck is the amount of Random Access Memory (RAM) available for loading the genome index.
Table 1: Recommended Hardware Specifications for STAR
| Component | Minimum Recommendation | Optimal Recommendation | Notes |
|---|---|---|---|
| CPU Cores | 4 cores | 8-16+ cores | STAR can utilize multiple threads for parallel processing. |
| RAM | 32 GB | 64 GB - 256 GB | Critical. Must hold the genome index in memory. For mammalian genomes (e.g., human, mouse), 32GB is the absolute minimum; 64GB is recommended for safe operation. |
| Storage (SSD) | 500 GB | 1-4 TB NVMe SSD | Fast I/O is essential. A high-speed SSD is required for the reference genome, indices, and interim files. |
| Operating System | 64-bit Linux (Ubuntu 20.04/22.04, CentOS 7/8) | 64-bit Linux or macOS | STAR is primarily developed for UNIX-like systems. Windows requires WSL2 or a virtual machine. |
Software Dependencies
A functional computational environment requires the following software to be installed and available in the system's PATH.
Table 2: Essential Software Dependencies
| Software | Minimum Version | Function & Role in Workflow |
|---|---|---|
| STAR | 2.7.10a | The core alignment software for mapping RNA-seq reads to a reference genome. |
| SAMtools | 1.15 | Utilities for manipulating SAM/BAM alignment files (sorting, indexing, conversion). |
| GCC/G++ | 7.0.0 | Compiler required to build STAR from source. |
| Git | 2.0+ | For cloning the STAR repository from GitHub. |
| Python | 3.8+ | For running auxiliary data processing and quality control scripts. |
| FastQC | 0.11.9 | Quality control tool for raw sequencing data (pre-alignment check). |
| Curl/wget | Latest | Command-line tools for downloading reference genomes and annotation files. |
Protocols
Protocol 1: System Preparation and Dependency Installation
Objective: To establish a clean computational environment with all required dependencies on a fresh Ubuntu 22.04 LTS installation.
- Update System Packages:
- Install Core Compilers and Tools:
- Install SAMtools and HTSlib (from source for latest features):
- Install Python and pip:
- Install FastQC:
Protocol 2: Building and Installing STAR from Source
Objective: To compile and install the latest version of the STAR aligner for optimal performance.
- Clone the STAR Repository:
- Navigate to Source Directory and Build:
- Add STAR to System PATH:
Verify installation:
STAR --version
Protocol 3: Generating a STAR Genome Index
Objective: To create a genome index for Homo sapiens (GRCh38.p14) required for alignment.
- Download Reference Genome and Annotation:
- Create Directory for Genome Index:
- Run STAR Genome Index Generation:
Parameters:
--runThreadN: Number of CPU threads.--sjdbOverhang: Read length minus 1.--genomeSAindexNbases: For small genomes, reduce this value (default 14).
Protocol 4: Performing Read Alignment with STAR
Objective: To align paired-end RNA-seq FASTQ files to the prepared genome index.
- Prepare Input and Output Directories:
Assume paired-end read files:
sample_1.fastq.gz,sample_2.fastq.gz. - Execute STAR Alignment:
Parameters:
--readFilesCommand zcat: For gzipped input.--outSAMtype BAM SortedByCoordinate: Outputs a sorted BAM file.--quantMode GeneCounts: Outputs read counts per gene.
Visualizations
STAR RNA-seq Alignment Setup Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Materials for RNA-seq Alignment
| Item/Category | Function/Explanation |
|---|---|
| Reference Genome (FASTA) | The nucleotide sequence of the target organism (e.g., human GRCh38). Serves as the map for aligning sequencing reads. |
| Gene Annotation (GTF/GFF) | File specifying genomic coordinates of genes, exons, splice junctions, and other features. Critical for splice-aware alignment and quantification. |
| STAR Genome Index | A pre-processed, binary representation of the genome optimized for ultra-fast searching. Generated once per genome/annotation version and loaded into RAM. |
| High-Quality RNA-seq Reads (FASTQ) | The raw input data. Must undergo quality control (e.g., FastQC) to ensure reliable alignment and downstream analysis. |
| Alignment Output (Sorted BAM) | The primary result file. Contains the genomic coordinates for each input read, used for visualization and further analysis (e.g., variant calling). |
| Gene Count Matrix | Tabular output from STAR's --quantMode. Represents the number of reads mapped to each gene, forming the basis for differential expression analysis. |
Step-by-Step STAR Protocol: From Genome Indexing to Final BAM Output
Within the thesis framework "How to perform read alignment with STAR for RNA-seq research," the initial and most critical computational step is generating a genome index. This pre-processing phase creates a searchable reference from a genome sequence and annotation file, enabling the ultrafast alignment performance of the STAR (Spliced Transcripts Alignment to a Reference) aligner. The quality of the index directly impacts alignment accuracy, speed, and splice junction discovery. These Application Notes detail the command parameters and best practices for this foundational phase.
Core Command Parameters and Quantitative Specifications
The basic command for generating a STAR genome index is STAR --runMode genomeGenerate. The following table summarizes the essential and advanced parameters, with quantitative guidance derived from current best practices and the STAR manual.
Table 1: Essential Parameters for STAR Genome Index Generation
| Parameter | Description | Typical Value / Recommendation | Rationale |
|---|---|---|---|
--genomeDir |
Path to the directory where the genome index will be stored. | User-defined directory path | The directory must be created prior to the run and writable. |
--genomeFastaFiles |
Path(s) to the FASTA file(s) of the reference genome. | e.g., Homo_sapiens.GRCh38.dna.primary_assembly.fa |
Provides the reference nucleotide sequences. Use a primary assembly, not a "toplevel" file with many patches. |
--sjdbGTFfile |
Path to the annotation file in GTF format. | e.g., Homo_sapiens.GRCh38.110.gtf |
Provides gene models for annotating junctions and improving mapping accuracy for spliced reads. |
--sjdbOverhang |
Length of the genomic sequence around annotated junctions to be used in constructing the splice junction database. | Read length minus 1. (e.g., 99 for 100bp paired-end reads). | Optimized value ensures reliable mapping of junction-spanning reads. The default (100) works for most read lengths. |
--runThreadN |
Number of threads to use for index generation. | 4-8, depending on available CPU cores. | Parallelizes the process, significantly reducing runtime. |
Table 2: Key Advanced Parameters for Performance and Resources
| Parameter | Description | Recommended Setting for Mammalian Genomes | Impact |
|---|---|---|---|
--genomeSAindexNbases |
Length of the SA (Suffix Array) pre-index "word". | 14 for genomes ~3 billion bases. |
Must be scaled down for smaller genomes: min(14, log2(GenomeLength)/2 - 1). Critical for memory usage. |
--genomeChrBinNbits |
Determines bin size for genome storage in memory. | 18 for genomes ~3 billion bases. |
Scale down for genomes with many small scaffolds (e.g., min(18, log2(GenomeLength/NumberOfScaffolds))). |
--limitGenomeGenerateRAM |
Maximum available RAM (bytes) for index generation. | e.g., 60000000000 (60GB) |
Must be set if system RAM is limited. The index process requires ~40GB for human/mouse. |
Detailed Protocol for Index Generation
Protocol 1: Generating a Genome Index for a Human RNA-seq Experiment
Objective: To create a STAR-compatible genome index from the human GRCh38 reference genome and Ensembl annotation for 100bp paired-end read alignment.
Materials & Pre-Processing:
- Reference Genome FASTA: Download the primary assembly (e.g.,
Homo_sapiens.GRCh38.dna.primary_assembly.fa.gzfrom Ensembl). - Annotation GTF: Download the corresponding GTF file (e.g.,
Homo_sapiens.GRCh38.110.gtf.gzfrom Ensembl). - Compute Environment: A Linux server or cluster node with minimum 32GB RAM (60GB recommended), 8+ CPU cores, and adequate storage (~30GB for the final index).
Methodology:
- Data Preparation:
Index Generation Command: Execute the STAR command with optimized parameters. Adjust
--runThreadNand--limitGenomeGenerateRAMaccording to your system.Verification: Upon successful completion, the
--genomeDirwill contain numerous files (e.g.,Genome,SA,SAindex,chrLength.txt,chrName.txt,chrStart.txt,genomeParameters.txt). Check the log file (Log.out) for any warnings and to confirm the process finished without error.
Visualization of Workflow and Logic
Diagram Title: STAR Genome Index Generation Workflow
Diagram Title: Logic for Determining Critical Index Parameters
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Materials for STAR Indexing
| Item | Function & Rationale | Example Source/Format |
|---|---|---|
| Reference Genome (FASTA) | The canonical DNA sequence of the organism against which RNA-seq reads are aligned. Using a "primary assembly" without haplotypes and patches reduces complexity and resource usage. | ENSEMBL (*.dna.primary_assembly.fa), UCSC, GENCODE |
| Gene Annotation (GTF/GFF3) | File containing coordinates of known genes, transcripts, exons, and other features. Crucial for STAR to build a database of known splice junctions, dramatically improving alignment accuracy within transcribed regions. | ENSEMBL (*.gtf), GENCODE (*.gtf), RefSeq |
| STAR Aligner Software | The aligner executable itself. Using an updated version ensures access to the latest features, optimizations, and bug fixes. | GitHub Releases (https://github.com/alexdobin/STAR/releases) |
| High-Performance Compute (HPC) Node | Index generation is computationally intensive, requiring significant RAM (~40GB for human) and multiple CPU cores for a timely completion. | Local server, institutional cluster, or cloud compute instance (e.g., AWS r6i.2xlarge). |
| Package/Environment Manager | Tool to ensure reproducible installation of STAR and its dependencies, avoiding version conflicts. | Conda, Bioconda, Docker, Singularity |
Application Notes
The two-pass mapping strategy in STAR is a powerful method for increasing the sensitivity of novel splice junction discovery in RNA-seq experiments. In standard one-pass alignment, STAR uses a supplied reference genome annotation (GTF/GFF file) to inform alignment and initially identify junctions. This approach can miss junctions not present in the annotation. The two-pass method addresses this by using the sample's own data to build a novel, sample-specific splice junction database, which is then used in a second alignment pass to improve mapping accuracy and novel junction detection.
Key Advantages:
- Enhanced Discovery: Significantly increases the number of uniquely mapped reads that span novel junctions.
- Sample-Specific Sensitivity: Crucial for studies involving non-canonical tissues, disease states with aberrant splicing, or poorly annotated genomes.
- Improved Quantification: Leads to more accurate transcript-level and gene-level quantification by correctly assigning reads spanning new junctions.
Quantitative Performance Summary:
Table 1: Typical Performance Gains with STAR Two-Pass Mode
| Metric | One-Pass Alignment | Two-Pass Alignment | Notes |
|---|---|---|---|
| Uniquely Mapped Reads | 80-85% | 82-87% | Modest increase; sample-dependent. |
| Novel Splice Junctions Detected | Baseline | 20-40% increase | Relative to junctions found in 1st pass. |
| Multi-mapped Reads | 5-10% | Slight decrease | Reassignment to novel junctions improves uniqueness. |
| Computational Resources | 1x | ~2x | Requires nearly double the runtime and temporary disk space. |
Detailed Experimental Protocols
Protocol 1: Basic Two-Pass Execution with STAR
This protocol details the step-by-step process for performing two-pass mapping on a single RNA-seq sample.
1. First Pass Alignment:
- Objective: Generate an initial alignment and extract a list of novel splice junctions.
- Command:
- Key Output:
sample_FirstPass_SJ.out.tab– a tab-separated file containing all detected splice junctions from this sample.
2. Junction Filtering and Compilation (for multiple samples):
- Objective: Combine junctions from multiple samples and filter for robust, novel junctions to create a new database.
- Command (to combine junctions from 3 samples):
- Filtering Recommendation: Use in-house scripts or tools to filter junctions based on:
- Uniquely Mapping Reads: Minimum threshold (e.g., ≥ 3 reads).
- Overhang: Minimum overhang length on both sides.
- Canonical vs. Non-canonical: Consider separate thresholds.
3. Second Pass Alignment:
- Objective: Re-align reads using the original genome annotation PLUS the novel junctions discovered in the first pass.
- Command:
- Key Parameters:
--sjdbFileChrStartEnd: Points to the combined/filtered junction file.--limitSjdbInsertNsj: Increases the limit for the number of junctions that can be inserted into the genome index.--quantMode GeneCounts: Outputs read counts per gene.--quantMode TranscriptomeSAM: Outputs a BAM aligned to the transcriptome for use with quantification tools like Salmon.
4. Index the Final BAM File:
Visualizations
Title: Two-Pass Mapping Workflow for Novel Junction Discovery
Title: Mapping Outcome Comparison: One-Pass vs. Two-Pass
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for STAR Two-Pass Mapping
| Item | Function & Purpose in Protocol |
|---|---|
| High-Quality Reference Genome (FASTA) | The DNA sequence of the organism under study. Required to build the initial STAR genome index. |
| Annotation File (GTF/GFF3) | Contains known gene models and splice junctions. Guides the first alignment pass and is augmented in the second pass. |
| STAR Aligner Software | The core splice-aware aligner capable of performing the two-pass mapping algorithm. |
| High-Performance Computing (HPC) Cluster or Server | Two-pass mapping is computationally intensive, requiring significant CPU, memory (~32GB+ RAM for mammalian genomes), and storage. |
| SAMtools | Software suite for processing and indexing the final sorted BAM alignment files. |
| Post-Alignment Quantification Tool (e.g., featureCounts, HTSeq) | To generate gene-level expression counts from the two-pass BAM file if not using STAR's built-in --quantMode. |
| Junction Filtering Script (Custom/Python/Perl/R) | To process the SJ.out.tab files from multiple samples, applying read count and overhang filters to create a robust novel junction list. |
| RNA-seq Data from Biological Replicates | Essential for distinguishing sample-specific technical artifacts from biologically reproducible novel splice junctions during filtering. |
Within the broader thesis on performing RNA-seq read alignment with STAR, command parameter selection is critical for balancing alignment speed, accuracy, and downstream analytical utility. This document provides detailed Application Notes and Protocols for essential and advanced STAR alignment parameters, focusing on output filtering and SAM/BAM file control. Proper configuration of these parameters directly impacts the quality of gene expression quantification and variant discovery.
Essential & Advanced Parameter Tables
Table 1: Core Output Filtering Parameters (--outFilterType)
| Parameter | Typical Values | Default | Function & Impact on Alignment |
|---|---|---|---|
--outFilterType |
Normal, BySJout |
Normal |
Normal: Standard filtering using alignment scores. BySJout: Uses information from previously discovered junctions (in the same sample) to guide filtering; reduces false novel junctions but is more stringent. |
--outFilterMultimapNmax |
Integer (e.g., 10, 20, 50) | 10 | Maximum number of loci a read is allowed to map to. Alignments exceeding this are considered multi-mapped and filtered out of the main BAM. Higher values retain more alignments but increase ambiguity. |
--outFilterMismatchNmax |
Integer (e.g., 5, 10) | 10 | Maximum number of mismatches per paired read (or unpaired read). For 2x100bp, default allows up to 10% mismatch rate. |
--outFilterMismatchNoverLmax |
Float (e.g., 0.04, 0.1) | 0.3 | Maximum mismatch ratio per read: mismatches / read length. More stringent than the absolute count. Setting to 0.04 allows only 4 mismatches per 100bp. |
--outFilterScoreMin |
Integer (e.g., 0, 10) | 0 | Minimum alignment score (sum of match scores - mismatch penalties). Positive values impose stringency. |
--outFilterIntronMotifs |
None, RemoveNoncanonical |
None |
RemoveNoncanonical: Filters out alignments containing non-canonical intron motifs (e.g., not GT-AG, GC-AG, AT-AC). Crucial for certain downstream analyses like pseudogene filtration. |
Table 2: SAM/BAM Output Control Parameters (--outSAMtype)
| Parameter | Values | Default | Function & Output Implications |
|---|---|---|---|
--outSAMtype |
None, BAM, SAM, BAM Unsorted, BAM SortedByCoordinate |
None (No SAM output) |
Defines the format and sorting of the main alignment output. BAM SortedByCoordinate is typically required for downstream tools (e.g., IGV, featureCounts). |
--outSAMmode |
None, Full |
None |
Full: Outputs all SAM attributes. Necessary for most downstream analyses. |
--outSAMattributes |
List (e.g., Standard, NH HI AS nM) |
Standard |
Controls which SAM tags are included. Standard = basic set. NH (Number of reported alignments) and HI (Hit index) are critical for multi-mappers. AS (Alignment score) and nM (Number of mismatches) are useful for QC. |
--outSAMunmapped |
None, Within |
None |
Within: Includes unmapped reads in the SAM/BAM output. Useful for assessing total reads. |
--outSAMprimaryFlag |
OneBestScore, AllBestScore |
OneBestScore |
Determines how the primary alignment (flag 0x100) is assigned. OneBestScore: one random best. AllBestScore: all top-scoring alignments are marked as primary (non-unique mapping). |
--outBAMcompression |
Integer (0-10) | 1 | BAM compression level. 0 = no compression, 10 = maximum (slowest). |
Experimental Protocols
Protocol 1: Optimizing Alignment Stringency for Differential Expression Analysis
Objective: Generate alignments with high confidence in uniquely mapped reads for accurate gene-level quantification.
- STAR Command:
- Rationale:
--outFilterType BySJoutand--outFilterMultimapNmax 1ensure only unique, junction-informed alignments are output for counting.--outFilterMismatchNoverLmax 0.04enforces high per-base accuracy.
Protocol 2: Permissive Alignment for Novel Junction and Variant Discovery
Objective: Maximize sensitivity for detecting novel splicing events and sequence variants, retaining multi-mapped reads for context.
- STAR Command:
- Rationale: Higher
--outFilterMultimapNmax, default mismatch ratio, and--outSAMattributes Allretain comprehensive mapping information.--twopassMode Basicenhances novel junction discovery.
Visualizations
Title: STAR Alignment Parameter Control Flow for RNA-seq
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in STAR RNA-seq Alignment |
|---|---|
| Reference Genome FASTA | The DNA sequence of the target organism. Provides the primary sequence against which reads are aligned. Must be consistent with the annotation. |
| Annotation File (GTF/GFF) | Defines genomic features (genes, exons, transcripts). Used by STAR for splice-aware alignment and read counting (--quantMode). |
| High-Quality RNA-seq Reads (FASTQ) | The input sequence data. Read length and quality (Phred scores) directly impact alignment rates and parameter choice (e.g., --outFilterMismatchNoverLmax). |
| STAR Index | A pre-processed genome file generated by STAR --runMode genomeGenerate. Contains the transformed reference for ultra-fast searching. Critical for performance. |
| Computational Resources (CPU/RAM) | STAR is resource-intensive. ≥ 32GB RAM and multiple cores are typically required for mammalian genomes. Impacts --runThreadN and overall runtime. |
| SAM/BAM Processing Tools (samtools) | Used to index, view, and manipulate alignment outputs from STAR (--outSAMtype). Essential for QC and preparing files for downstream analysis. |
| Read Counting Software (featureCounts, HTSeq) | Downstream tools that use the coordinate-sorted BAM (--outSAMtype BAM SortedByCoordinate) and gene annotation to generate expression matrices. |
Within the broader thesis on performing read alignment with STAR for RNA-seq research, the critical step following a successful run is the interpretation of the log file. The final log file (Log.final.out) provides a comprehensive summary of the alignment process and is essential for assessing data quality, identifying potential issues, and ensuring the integrity of downstream analyses. This document details the key metrics, their biological and technical significance, and protocols for their evaluation.
Key Metrics: Definitions and Interpretation
The STAR log file is organized into distinct sections. The following tables summarize the crucial metrics for quality assessment.
| Metric | Description | Ideal Range/Interpretation | Potential Issue Indicator |
|---|---|---|---|
| Number of input reads | Total reads processed from the FASTQ file. | Matches sequencing output. | Discrepancy suggests file corruption or incorrect input. |
| Uniquely mapped reads % | Percentage of reads mapped to a single genomic location. | Typically >70-80% for human/mouse. Higher is better. | Low % suggests poor RNA quality, high fragmentation, or contamination. |
| % of reads mapped to multiple loci | Reads aligned to 2-10 locations. | Varies; 10-30% common for RNA-seq due to paralogs. | Very high % may indicate high repeat content or poor genome annotation. |
| % of reads mapped to too many loci | Reads aligned to >10 locations. | Should be low (<5-10%). | High % suggests reads from repetitive elements (e.g., LINE, SINE). |
| % of reads unmapped: too short | Reads trimmed below minimum length for alignment. | Should be minimal (<1%). | High % indicates adapter contamination or excessive trimming. |
| % of reads unmapped: other | Reads failing to align for other reasons. | Should be very low. | May indicate sequencing errors or novel sequences not in genome. |
Table 2: Splice Junction and Insertion/Deletion Metrics
| Metric | Description | Significance for RNA-seq |
|---|---|---|
| Number of splices: Total | Total splice junctions detected in all aligned reads. | Indicator of transcriptome complexity. |
| Number of splices: Annotated (sjdb) | Splice junctions matching those in the supplied annotation (GTF). | High % (>80-90%) suggests alignment agrees with known annotation. |
| Number of splices: Novel | Splice junctions not in the supplied annotation. | Source of potential novel isoform discovery. Requires validation. |
| Deletion rate / Insertion rate | Frequency of small indels in aligned reads. | Should be very low (~0.01-0.1%). High rates may indicate sequencing errors or poor reference quality. |
| Mismatch rate per base, % | Non-indel mismatch frequency. | ~0.5-2%. Elevated rates can indicate sequence diversity (e.g., polymorphisms) or quality issues. |
Experimental Protocol: Quality Assessment Workflow
Protocol 1: Systematic Log File Evaluation for RNA-seq Alignment
Objective: To assess the quality of a STAR alignment run using the Log.final.out file.
Materials & Equipment:
- STAR
Log.final.outfile from alignment job. - Reference genome assembly name and version.
- RNA-seq library preparation kit documentation (e.g., strandedness).
- Sample metadata (e.g., tissue type, known quality issues).
Procedure:
- File Inspection: Open the
Log.final.outfile in a text editor. Verify it is the final log (not the progress logLog.progress.out). - Check Completion: Confirm the log ends with "Finished successfully". Any early termination indicates a critical error.
- Input/Output Verification:
- Compare "Number of input reads" with the output of
fastqcor raw sequence counts. - Ensure "Average input read length" matches library preparation expectations.
- Compare "Number of input reads" with the output of
- Primary Alignment Assessment:
- Record "Uniquely mapped reads %". Compare to historical runs of similar sample types. A drop >10% warrants investigation.
- Evaluate multi-mapping reads ("% of reads mapped to multiple loci"). Context is sample-dependent.
- Splice Junction Analysis:
- Calculate the ratio:
(Annotated splices / Total splices). A low ratio (<70%) in a well-annotated organism suggests potential novel splicing or alignment artifacts. - Note the number of novel splices for downstream exploratory analysis.
- Calculate the ratio:
- Error Rate Scrutiny:
- Examine "Mismatch rate per base, %" and "Deletion/Insertion rate". Compare to expected rates for your sequencing platform.
- Strandedness Verification (if applicable):
- For stranded libraries, check "% of reads mapped to opposite strand". This should be very low (<5%) for correctly specified
--outSAMstrandFieldparameter.
- For stranded libraries, check "% of reads mapped to opposite strand". This should be very low (<5%) for correctly specified
- Documentation: Compile all key metrics into a quality control table for the project.
Visualization: STAR Alignment Assessment Workflow
Diagram Title: STAR Log File Assessment Decision Workflow
The Scientist's Toolkit: Essential Reagents & Solutions
Table 3: Key Research Reagent Solutions for STAR Alignment & QC
| Item | Function/Brief Explanation |
|---|---|
| High-Quality Reference Genome (FASTA) | The genomic sequence to which reads are aligned. Must match organism and be contiguously assembled. |
| Annotation File (GTF/GFF3) | Gene transfer format file defining known gene models, transcripts, and exon boundaries. Crucial for splice-aware alignment and junction database (--sjdbGTFfile). |
| STAR Aligner Software | Spliced Transcripts Alignment to a Reference. Executable software for performing the alignment. |
| High-Performance Computing (HPC) Cluster or Server | STAR is memory-intensive (~30+ GB for mammalian genomes). Adequate RAM and multi-core CPUs are essential. |
| RNA-seq FASTQ Files | The raw sequencing reads in standard format. Should be demultiplexed and quality-checked prior to alignment. |
| FASTQC/MultiQC Tool | For initial quality control of FASTQ files and aggregated reporting of STAR logs across multiple samples. |
| SAM/BAM Tools (e.g., samtools) | For processing, indexing, and manipulating the sequence alignment/map (SAM/BAM) output files from STAR. |
| RSeQC or Qualimap | Tool for comprehensive quality control of RNA-seq alignment data, building upon STAR's log metrics. |
Within the context of a STAR-aligned RNA-seq experiment, the generation of a Sequence Alignment/Map (SAM) file is only the first step. The resulting SAM file, while containing all alignment information, is unsorted, highly redundant, and not optimized for downstream analysis. Post-alignment processing—specifically sorting, deduplication (if required), and indexing—transforms this raw alignment into a compressed, query-ready Binary Alignment/Map (BAM) file. This processed file is the critical input for subsequent quantification (e.g., via featureCounts), variant calling, and visualization. This protocol details the essential steps to convert a STAR output SAM into a final, analysis-ready BAM file.
Core Concepts and Workflow
The standard post-alignment pipeline involves three sequential steps: Sorting, Deduplication (optional for RNA-seq), and Indexing. Sorting orders alignments by their genomic coordinates, which is mandatory for many downstream tools and for efficient indexing. Deduplication removes PCR artifacts, which is more common in DNA-seq but can be applied to RNA-seq in specific experimental designs (e.g., single-cell or unique molecular identifier (UMI)-based assays). Finally, indexing creates a separate, small file that allows rapid random access to alignments in any genomic region, enabling fast visualization in browsers like IGV and efficient counting by region-based tools.
The following diagram illustrates the logical workflow from STAR alignment to a ready-to-use BAM file.
Diagram Title: Post-Alignment BAM Processing Workflow for RNA-seq
Detailed Experimental Protocols
Protocol 3.1: Sorting and Compressing SAM to BAM usingsamtools
Purpose: Convert the uncompressed, coordinate-unsorted SAM file into a compressed, coordinate-sorted BAM file. Reagents & Tools: Samtools suite (v1.19+ recommended), sufficient disk space (BAM is ~40% of SAM size).
- Prerequisite: Ensure
samtoolsis installed and in yourPATH. Verify STAR alignment completed successfully. - Command:
-@ 8: Use 8 CPU threads for sorting.-o: Specifies the output sorted BAM file name.Aligned.out.sam: The default SAM output from STAR.
- Validation: Check the BAM header to confirm sort order:
Expected output:
@HD VN:1.6 SO:coordinate.
Protocol 3.2: Marking Duplicates usingsamtools markdup(Optional)
Purpose: Identify and flag potential PCR duplicate reads. Note: Not routinely recommended for standard bulk RNA-seq but critical for UMI protocols. Reagents & Tools: Samtools, Picard Tools (alternative).
- Pre-requisite: Sorting must be complete. For
samtools markdup, first need to identify duplicates. - Step 1 - Add Read Group Tags (if not from STAR): Some downstream tools require
@RGheader. - Step 2 - Identify and Mark Duplicates:
-sinmarkdup: Print basic statistics.
- Output: A BAM where duplicate reads are flagged with bit 0x400.
Protocol 3.3: Indexing the Final BAM File usingsamtools index
Purpose: Generate a .bai index file for rapid random access to the BAM file.
Reagents & Tools: Samtools.
- Prerequisite: The BAM file must be coordinate-sorted.
- Command:
This creates
Aligned.final.markdup.bam.bai. - Verification: Use a quick query to test index functionality:
Protocol 3.4: Quality Control and Integrity Checking
Purpose: Assess the quality and completeness of the post-alignment processing. Reagents & Tools: Samtools, FastQC (optional re-run).
- Generate Alignment Statistics:
- Verify BAM Integrity: Ensure the file is not truncated.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Relevance in Post-Alignment Processing |
|---|---|
| Samtools | Core software suite for manipulating SAM/BAM files. Used for view, sort, index, flagstat, and markdup operations. Essential for all steps. |
| High-Performance Computing (HPC) Cluster | Sorting and indexing are memory and CPU-intensive. A cluster or server with adequate RAM (>16GB for mammalian genomes) and multiple cores significantly speeds up processing. |
| Picard Tools (MarkDuplicates) | A common alternative to samtools markdup for duplicate marking, often cited in GATK best practices pipelines. Provides detailed duplicate metrics. |
| UMI-Tools | If the RNA-seq library was prepared with Unique Molecular Identifiers, this specialized toolset is required for accurate deduplication based on UMI sequences, not just genomic coordinates. |
| Integrative Genomics Viewer (IGV) | Visualization tool that relies on an indexed BAM (.bai) file to enable real-time browsing of alignments across the genome, used for final QC of the processed BAM. |
| RNA-seq Quantification Tool (e.g., featureCounts, HTSeq) | The primary downstream application for a ready-to-use BAM file. These tools require coordinate-sorted, indexed BAMs to efficiently count reads overlapping genomic features. |
Table 1: Comparative Output Metrics from Post-Alignment Processing Steps (Example Human RNA-seq Dataset)
| Processing Step | File Type | Approximate Size (GB) | Key Content/Change | Required for Downstream? |
|---|---|---|---|---|
| STAR Output | SAM (Aligned.out.sam) | 120.0 | All alignments, unsorted, uncompressed. | No - Intermediate format. |
| After Sorting | BAM (Aligned.sortedByCoord.out.bam) | 45.0 | All alignments, coordinate-sorted, compressed. | Yes - Input for indexing/deduplication. |
| After Deduplication (Optional) | BAM (Aligned.final.markdup.bam) | 42.0 | Duplicate reads flagged (not removed). | Context-dependent. |
| Final Index | BAI Index File (Aligned.final.markdup.bam.bai) | 0.002 | Binary index of genomic positions. | Yes - For visualization & counting. |
Table 2: Common samtools flagstat Metrics for a Processed BAM File
| Metric | Example Count | Interpretation |
|---|---|---|
| Total reads (QC-passed) | 45,678,123 | Total number of reads in the final BAM. |
| Mapped reads | 43,394,216 (94.9%) | Percentage successfully aligned by STAR. |
| Reads marked as duplicate | 2,100,456 (4.6%) | Estimate of PCR duplicates (if deduplication run). |
| Reads paired in sequencing | 45,678,123 | Confirms paired-end data. |
| Properly paired reads | 41,123,987 (90.0%) | Both pairs mapped correctly relative to each other. |
Solving Common STAR Alignment Issues and Maximizing Performance
Within the context of RNA-seq analysis using STAR (Spliced Transcripts Alignment to a Reference), a low overall alignment rate is a critical quality control failure point. It indicates a substantial portion of sequenced reads could not be placed in the reference genome, leading to data loss, reduced statistical power, and potentially biased downstream results. This Application Note details the systematic diagnosis of this issue, its root causes, and validated solutions.
Primary Causes and Quantitative Impact
Low alignment rates typically stem from three core areas: sample/library quality, reference mismatch, and STAR parameter configuration.
Table 1: Common Causes of Low Alignment Rates with STAR
| Category | Specific Cause | Typical Alignment Rate Impact | Key Diagnostic Indicator |
|---|---|---|---|
| Sample/Library Quality | RNA Degradation | 10-40% reduction | Low RNA Integrity Number (RIN < 7) |
| Adapter Contamination | 15-50% reduction | High % of unmapped reads due to 'other' | |
| PCR Duplicates (Extreme Over-amplification) | 5-20% reduction (via complexity loss) | Exceptionally high duplication rates (>80%) | |
| Low Sequencing Quality | Variable reduction | High % of unmapped reads with low mapping quality (MAPQ=0) | |
| Reference Mismatch | Incorrect Genome Build/Version | 50-90% reduction | Consistently low % of uniquely mapped reads |
| Contaminated Sample (e.g., host-pathogen) | 20-70% reduction | High % of reads mapping to unexpected genomes | |
| High Genetic Divergence from Reference | 20-60% reduction | Low % of uniquely mapped, high multi-mapping | |
| STAR Parameters | Insufficient --outFilterScoreMinOverLread |
<5% change | Slight increase in uniquely mapped reads if adjusted |
Incorrect --sjdbOverhang (for annotated splice junctions) |
5-15% reduction for spliced reads | Low % of reads mapped to splice junctions | |
Overly Stringent --outFilterMismatchNmax or --outFilterMultimapNmax |
10-30% reduction | High % of unmapped reads: 'too many mismatches' or 'too many loci' |
Diagnostic Protocol
Follow this sequential workflow to identify the cause of low alignment.
Diagram 1: Diagnostic Workflow for Low STAR Alignment.
Protocol: Initial Quality Control
Objective: Assess raw read quality and adapter contamination.
- Tool: FastQC (v0.12.1) and MultiQC (v1.21).
- Command:
- Interpretation: Open the MultiQC HTML report. Note:
- Per base sequence quality: Scores below 20 in later cycles indicate quality drop.
- Adapter content: Any adapter contamination >1% requires trimming.
- Sequence duplication levels: Critically high levels suggest low library complexity.
Protocol: Inspect STAR Log File
Objective: Parse STAR's summary statistics to categorize unmapped reads.
- File:
Log.final.outfrom the failed alignment run. - Key Sections:
- Uniquely mapped reads %: The primary metric of interest.
- % of reads unmapped: other: High percentage suggests adapter, quality, or too-short reads post-clipping.
- % of reads unmapped: too many mismatches: Indicates potential reference mismatch or overly stringent
--outFilterMismatchNmax. - % of reads unmapped: too short: Reads were trimmed below minimum length (
--outFilterScoreMinor adapter removal). - % of reads unmapped: too many loci: Reads map to too many places, suggesting repetitive sequences or low
--outFilterMultimapNmax.
Protocol: Validate Reference Compatibility
Objective: Confirm the reference genome matches the sample species and strain.
- Check the genome fasta and annotation GTF files used in STAR genome generation.
- For genetically diverse samples (e.g., non-model organisms, patient-derived xenografts), consider the need for a customized reference.
- For suspected contamination, perform a quick alignment to a combined reference (e.g., host+pathogen) using a subset of reads.
Solutions and Optimization Protocols
Solution Protocol: Adapter Trimming and Quality Filtering
Objective: Remove technical sequences and low-quality bases.
- Tool: fastp (v0.23.4) or Trimmomatic (v0.39).
- fastp Command (Recommended):
- Re-align the trimmed FASTQ files with STAR.
Solution Protocol: STAR Parameter Optimization
Objective: Adjust alignment sensitivity and stringency. Table 2: Key STAR Parameters for Troubleshooting Low Alignment
| Parameter | Default | Suggested Adjustment for Low Rates | Rationale |
|---|---|---|---|
--outFilterScoreMinOverLread |
0.66 | Reduce to 0.5 or 0.4 | Relaxes the minimal alignment score per read length, allowing more alignments. |
--outFilterMatchNminOverLread |
0.66 | Reduce to 0.5 or 0.4 | Relaxes the minimal matched bases per read length. |
--outFilterMismatchNmax |
10 | Increase to 15 or 20 | Allows more mismatches, helpful for divergent samples. |
--outFilterMultimapNmax |
10 | Increase to 50 or 100 | Allows reads mapping to more locations before being classified as "unmapped: too many loci". |
--alignSJDBoverhangMin |
5 | Reduce to 3 (minimum is 2) | Reduces the minimum overhang for annotated spliced alignments, aiding short exon mapping. |
--seedSearchStartLmax |
50 | Reduce to 30 | Speeds up alignment; can marginally improve sensitivity for some reads. |
--outReadsUnmapped Fastx |
None | Add --outReadsUnmapped Fastx |
Outputs unmapped reads for further analysis (e.g., contamination check). |
Optimization Command Example:
Solution Protocol: Using a Modified Reference
Objective: Align to the correct genome or a pan-genome.
- For contaminations: Create a combined reference (e.g., human + mouse, host + virus).
- For non-model organisms: Use a closely related species or de novo assembly if available.
- For personalized medicine: Use patient-specific genome if variants are known.
- Regenerate the STAR genome index with the new reference fasta and GTF files before alignment.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for High-Quality STAR Alignment
| Item | Function/Application | Example Product/Kit |
|---|---|---|
| High-Quality RNA Isolation Kit | Ensures high-integrity, non-degraded input RNA, the single most important factor for successful RNA-seq. | QIAGEN RNeasy Plus Mini Kit, Zymo Research Direct-zol RNA Miniprep. |
| Strand-Specific RNA Library Prep Kit | Creates sequencing libraries that preserve strand-of-origin information, crucial for accurate transcript quantification. | Illumina Stranded mRNA Prep, NEBNext Ultra II Directional RNA Library Prep Kit. |
| RNA Integrity Assay | Quantitatively assesses RNA degradation before library prep. Accept only RIN > 8 for optimal results. | Agilent RNA 6000 Nano Kit (Bioanalyzer). |
| Dual-Size Selection Beads | Precisely sizes library fragments, removing adapter dimers and overly large fragments which harm alignment. | SPRIselect Beads (Beckman Coulter), AMPure XP Beads. |
| Polymerase with High Fidelity | Used in cDNA amplification during library prep to minimize PCR errors that create mismatches. | Q5 High-Fidelity DNA Polymerase (NEB), KAPA HiFi HotStart ReadyMix. |
| Low-Bind Tubes and Tips | Prevents loss of low-concentration nucleic acids during library preparation and QC steps. | Eppendorf LoBind tubes, Axygen Low-Retention tips. |
| Commercial STAR-Aligned Benchmarks | Control RNA-seq samples (e.g., from SEQC/MAQC consortium) with known expected alignment rates to validate pipeline performance. | Horizon Discovery Multiplex ICF RNA Spike-in Controls (used post-alignment for QC). |
Within the broader thesis on performing RNA-seq read alignment with STAR, managing computational resources becomes paramount when dealing with large genomes (e.g., mammalian, plant) or large-scale experiments (e.g., multi-sample cohorts, single-cell RNA-seq). STAR's performance is highly dependent on parameter selection, which directly influences memory (RAM) consumption, runtime, and disk usage. This protocol details strategies for parameter tuning to optimize efficiency without compromising alignment accuracy.
STAR's alignment process occurs in two main phases: Genome Loading and Read Alignment. Key parameters affecting memory and runtime are associated with each.
Table 1: Key STAR Parameters for Resource Management
| Parameter | Default Value | Impact on Memory | Impact on Runtime | Recommendation for Large Genomes/Experiments |
|---|---|---|---|---|
--genomeSAindexNbases |
14 | High: Defines genome index size. Too low for large genome → huge memory; too high → slow. | High: Affects indexing and alignment speed. | For large genomes (>3Gb), use min(14, log2(GenomeLength)/2 - 1). For human (3.3Gb), use 14. |
--genomeSAsparseD |
1 | High: Controls sparsity of SA index. Increasing reduces memory. | Medium: Slightly increases runtime. | Use 2 for very large genomes to reduce RAM (e.g., >4GB genome). |
--limitGenomeGenerateRAM |
31000000000 | Critical: Limits RAM (in bytes) for genome generation. | - | Set to available physical RAM for index generation. |
--limitIObufferSize |
150000000 | Medium: Size of input/output buffers. | Low | Can be reduced to ~30,000,000 if RAM is limited. |
--limitOutSJoneRead |
1000 | Low | Low | Keep default to avoid missing splice junctions. |
--limitOutSJcollapsed |
1000000 | Medium: Limits RAM for collapsed splice junctions. | Low | Increase for large samples (>200M reads). |
--limitBAMsortRAM |
0 | High: Limits RAM for BAM sorting. If 0, uses --limitGenomeGenerateRAM. |
Medium | Set explicitly to available RAM if BAM sorting fails. |
--outBAMsortingBinsN |
50 | Low | Medium: More bins can reduce sorting time. | Increase to 100 or more for very large BAM files. |
--runThreadN |
1 | Low | Critical: Parallel processing. | Set to the number of available CPU cores. Major runtime reduction. |
--readFilesCommand |
- | Low | Medium | For compressed .gz files, use zcat or gzip -c to reduce disk I/O time. |
--outFilterScoreMinOverLread & --outFilterMatchNminOverLread |
0.66 & 0.66 | Low | High: Stricter filters reduce output size and downstream time. | Loosen (e.g., 0.33) for noisy data or damaged RNA to maintain sensitivity. |
--outSAMtype |
SAM | High: BAM sorted output requires more RAM for sorting. | Medium: Sorting adds time. | Use BAM Unsorted or BAM SortedByCoordinate with sufficient RAM. SortedByCoordinate is standard. |
--outWigType |
None | High: Wiggle file generation requires significant RAM. | High | Avoid or generate per-read-length wig files only if needed. |
--alignSJDBoverhangMin |
5 | Low | Low | Keep default for accuracy. |
--quantMode |
None | Medium: Gene counting adds compute. | High | Use --quantMode GeneCounts for direct counting, saving separate counting step. |
Diagram Title: Resource Impact of STAR Parameter Categories
Experimental Protocols
Protocol 3.1: Benchmarking STAR Parameters for a Large Genome
Objective: Systematically test key parameters to find the optimal balance between runtime, memory use, and mapping rate for a large genome (e.g., human, mouse).
Materials:
- Computing Cluster/Server with ≥ 32 GB RAM, 10+ CPU cores.
- Reference genome FASTA and GTF annotation files.
- Sample RNA-seq FASTQ files (≥ 20 million read pairs recommended).
- STAR software (v2.7.10a or later).
Procedure:
- Genome Indexing with Varied Parameters:
- Generate multiple genome indices in separate directories.
- Vary
--genomeSAindexNbases(e.g., 12, 14, 15 for human). - Vary
--genomeSAsparseD(e.g., 1, 2). - Use
--runThreadNwith your core count. - Command Template:
Alignment Benchmarking:
- Align the same sample FASTQ files using each generated index.
- Use a consistent alignment command, but vary one alignment parameter at a time (e.g.,
--outFilterScoreMinOverLread). - Record Peak Memory (Virt/Res), Wall Clock Time, CPU Time, and Uniquely Mapped Read % from the
Log.final.outfile. - Command Template:
Data Analysis:
- Compile results into a table (see Table 2 below).
- Plot runtime vs. memory vs. mapping rate.
- Select the parameter set that yields >90% unique mapping, within acceptable runtime, and under available RAM limits.
Table 2: Example Benchmark Results for Human Genome (20M Read Pairs)
Index (NBases/SparseD) |
Align Filter Score | Peak RAM (GB) | Wall Time (min) | CPU Time (hr) | % Unique Mapped |
|---|---|---|---|---|---|
| 14 / 1 | 0.66 | 28.5 | 42 | 8.1 | 92.5 |
| 14 / 2 | 0.66 | 24.1 | 48 | 9.2 | 92.3 |
| 15 / 1 | 0.66 | 32.8 | 38 | 7.8 | 92.6 |
| 14 / 1 | 0.33 | 28.7 | 40 | 7.9 | 93.1 |
| 14 / 1 | 0.80 | 28.1 | 45 | 8.5 | 89.8 |
Diagram Title: STAR Parameter Benchmarking Workflow
Protocol 3.2: Optimizing for a Multi-Sample Batch Experiment
Objective: Efficiently process hundreds of samples by minimizing per-sample runtime and managing parallel jobs.
Procedure:
- Shared Resource: Generate a single, optimized genome index (using Protocol 3.1 results) on a shared, high-speed storage drive.
- Parallelization Strategy:
- Use a cluster job scheduler (e.g., SLURM, SGE) to submit array jobs.
- Set
--runThreadNto 6-8 per job, balancing core usage across many concurrent jobs. - Critical: Limit the number of concurrent jobs based on available shared RAM (Total RAM / Peak RAM per job).
- Reduce I/O Bottlenecks:
- Use local node SSD scratch space for temporary files during alignment. Use STAR's
--outTmpDirflag. - Final BAM outputs should be written to a high-performance network storage.
- Use local node SSD scratch space for temporary files during alignment. Use STAR's
- Two-Pass Mode for Novel Junctions: If required, run 1st pass for all samples with
--twopassMode Basic, collecting new junctions. Then a 2nd pass using the merged SJ file.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Materials for Large-Scale STAR Alignment
| Item | Function & Relevance |
|---|---|
| High-Performance Computing (HPC) Cluster | Enables parallel processing of multiple samples simultaneously via job arrays. Essential for large cohorts. |
| Large-Memory Node (≥ 64 GB RAM) | Required for generating and loading the genome index of large organisms. Prevents crashes during alignment. |
| High-Speed Local Scratch Storage (NVMe/SSD) | Used for STAR's temporary files (--outTmpDir). Dramatically reduces I/O wait times during read alignment and BAM sorting. |
| Network-Attached Storage (NAS) with High IOPS | Stores raw FASTQ files, genome indices, and final BAM outputs. Requires high input/output operations per second (IOPS) for concurrent access by many cluster jobs. |
| STAR Aligner (v2.7.x+) | The core alignment software. Newer versions often contain optimizations for speed and memory. |
| SAMtools / BAMtools | For post-alignment processing, indexing, and quality checks of output BAM files. |
| RNA-seq QC Tool (e.g., FastQC, MultiQC) | To verify mapping quality and ensure parameter tuning has not introduced biases. |
| Cluster Job Scheduler (e.g., SLURM) | Manages resource allocation, queues, and submission of hundreds of alignment jobs efficiently. |
| Containerization (Docker/Singularity) | Provides a reproducible environment with all dependencies (STAR, tools) configured, ensuring consistency across runs. |
Addressing Multi-Mapping Reads and Ambiguous Alignments
Multi-mapping reads, which align to multiple genomic loci with equal or near-equal quality, are a significant challenge in RNA-seq analysis. In complex transcriptomes, 10-30% of reads can be multi-mapping, complicating accurate quantification. STAR (Spliced Transcripts Alignment to a Reference), by default, reports one alignment per read. The handling of these ambiguous alignments directly impacts downstream analyses such as isoform quantification and differential expression.
Table 1: Prevalence and Handling of Multi-Mapping Reads in STAR
| Parameter | Typical Value / Method | Impact on Quantification |
|---|---|---|
| Fraction of multi-mapping reads in total RNA-seq | 15-30% | High; can bias expression estimates if not addressed. |
STAR default output (--outSAMmultNmax) |
1 (single best alignment) | Unambiguous counts but potential loss of information from paralogous genes. |
--winAnchorMultimapNmax |
Default: 50 | Maximum number of loci anchors are allowed to map to. |
--outFilterMultimapNmax |
Default: 10 | Maximum number of alignments allowed for a read; reads exceeding this are considered unmapped. |
| Common post-alignment resolution tool | --quantMode with GeneCounts |
Assigns reads to genes based on supplied annotation (GTF). |
Experimental Protocols for Managing Ambiguity
Protocol 2.1: Basic STAR Alignment with Multi-Mapping Control
This protocol details the standard alignment step with key parameters for initial multi-mapping read management.
Materials:
- FASTQ files (read 1 and read 2 for paired-end).
- Reference genome index built by STAR.
- Gene annotation file in GTF format.
Method:
- Run STAR Alignment:
- Output Interpretation: The
sample_aligned.ReadsPerGene.out.tabfile contains counts where multi-mapping reads are assigned to a single gene based on the first matching feature in the GTF. Reads mapping to multiple genes are excluded from counts.
Protocol 2.2: Post-Hoc Resolution Using Expectation-Maximization (Salmon)
For more accurate quantification in the presence of multi-mapping reads, use a lightweight alignment/quantification tool like Salmon in its alignment-based mode.
Materials:
- STAR-aligned BAM file (from Protocol 2.1, run with
--outSAMmultNmax> 1 if using STAR's BAM as input). - Transcriptome FASTA file (transcripts, not genome).
- Salmon software.
Method:
- Generate Transcriptome BAM (Optional but recommended for STAR BAMs): Use
rsem-tbam2gbamif needed, or generate decoy-aware transcriptome. - Run Salmon Quantification:
- Mechanism: Salmon uses an expectation-maximization (EM) algorithm to probabilistically distribute multi-mapping reads across all potential loci of origin, weighted by the abundance of transcripts, providing more accurate estimates.
Protocol 2.3: Retaining All Alignments for Specialized Analysis
For analyses requiring all possible alignments (e.g., chimera detection, certain variant calling).
Method:
- Run STAR with Multi-Alignment Output:
- Process the BAM: The output BAM will contain multiple alignment records for a single read, distinguished by the
NH(number of reported alignments) andHI(alignment hit index) tags. Use tools likesamtoolswith care, as some may not be designed for multi-alignment BAMs.
Visualization of Decision Workflows
STAR Multi-Mapping Read Decision Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for Addressing Multi-Mapping Reads
| Tool / Reagent | Function | Key Consideration |
|---|---|---|
| STAR Aligner | Performs spliced alignment of RNA-seq reads to a reference genome. | Core tool; settings like --outFilterMultimapNmax and --outSAMmultNmax control initial multi-map reporting. |
| Salmon | Fast, bias-aware quantification of transcript expression using lightweight alignment & a rich statistical model. | Employs an EM algorithm to resolve multi-mapping reads probabilistically. Can use STAR BAM as input. |
| RSEM | (RNA-Seq by Expectation Maximization). Quantifies transcript abundances from aligned or unaligned data. | Includes a robust EM algorithm specifically designed for ambiguous read assignment. |
Picard Tools (FilterSamReads) |
Filters BAM files based on various criteria. | Can be used to isolate or remove reads based on the NH tag post-alignment. |
SAM/BAM Format with NH Tag |
Standard alignment output format. The NH tag indicates the number of reported alignments for a read. |
Essential for identifying multi-mapping reads in downstream processing. |
| High-Quality Annotated Reference (GTF/GFF) | File defining genomic coordinates of genes, transcripts, and exons. | Critical for accurate assignment of reads to features; incompleteness increases ambiguity. |
| Decoy-Aware Transcriptome | A combined reference of target transcripts and "decoys" (non-target genomic sequences). | Used by tools like Salmon to reduce false assignment of multi-mapping reads to homologous regions. |
Dealing with Strand-Specific Libraries and Annotation Mismatches
Within the broader thesis on performing RNA-seq read alignment with STAR (Spliced Transcripts Alignment to a Reference), managing strand-specific libraries and potential annotation mismatches is critical for accurate transcript quantification and downstream analysis. Strand-specific sequencing preserves the information of which genomic strand originated the RNA molecule, significantly improving the accuracy of novel transcript discovery and reducing antisense transcription artifacts. However, a mismatch between the library preparation protocol's strand orientation and the parameters used during alignment and quantification can lead to severe misinterpretation of gene expression, with counts assigned to the incorrect gene or strand.
Common Strand-Specific Library Protocols
Library preparation kits from various vendors produce different strand orientations. The critical parameter is the relationship between the first sequenced read (R1) and the original RNA strand.
Table 1: Common Strandedness Protocols and STAR Parameters
| Library Type | Description | R1 Orientation Relative to RNA | Common Kit Examples | STAR --outSAMstrandField |
FeatureCounts/HTSeq -s |
|---|---|---|---|---|---|
| Unstranded (Non-specific) | No strand information preserved. | N/A | Standard Illumina TruSeq (non-stranded) | intronMotif | no (0) |
| Forward (Sense) | R1 is complementary to the RNA. | Reverse (antisense to transcript) | Illumina TruSeq Stranded Total RNA, SMARTer Stranded | intronMotif | yes (1) |
| Reverse (Antisense) | R1 is identical to the RNA. | Forward (sense to transcript) | Illumina TruSeq Stranded mRNA, dUTP-based methods | intronMotif | reverse (2) |
Note: The dUTP method (e.g., Illumina TruSeq Stranded mRNA) is the most common, generating "reverse" stranded libraries.
Impact of Strand Mismatch on Quantification
Using incorrect strandedness parameters can lead to significant data loss or misassignment. The following table summarizes the typical effect when comparing correct vs. incorrect -s parameter in a quantification tool like featureCounts for a reverse-stranded library.
Table 2: Quantitative Impact of Strand Parameter Mismatch
| Quantification Scenario | % of Reads Assigned to Genes | % of Reads Assigned to Antisense of Genes | % of Reads Unassigned (No Feature) | Primary Consequence |
|---|---|---|---|---|
Correct (-s 2) |
70-85% | 2-5% | 15-25% | Accurate expression profile. |
Incorrect (-s 0) |
35-45% | 35-45% | 15-25% | Double-counting, inflated expression, antisense noise. |
Incorrect (-s 1) |
2-5% | 70-85% | 15-25% | Severe loss of signal; counts on wrong strand. |
Experimental Protocol: Determining Library Strandedness
If the library preparation method is unknown, an empirical verification protocol must be executed.
Protocol 3.1: Empirical Strandedness Verification using Infer Experiment (RSeQC Package)
Objective: Determine the strandedness of an RNA-seq library by comparing the alignment orientation of reads to known gene annotations.
Materials & Reagents:
- Aligned BAM File: From a preliminary STAR run using
--outSAMstrandField intronMotif. - Reference Gene Annotation: In BED12 format (can be converted from GTF using
gtf2bed). - RSeQC Software: Installed via pip (
pip install RSeQC). - Unix-based computing environment.
Procedure:
- Generate a BED12 file from your reference GTF annotation.
- Run the
infer_experiment.pyscript from the RSeQC package. - Interpret the output. The script will print results similar to:
- If "1++,1--,2+-,2-+" is the dominant fraction (e.g., >0.8), the library is reverse-stranded (dUTP method).
- If "1+-,1-+,2++,2--" is dominant, the library is forward-stranded.
- If ratios are close to 0.5 each, the library is unstranded.
Protocol 3.2: STAR Alignment with Correct Strandedness Parameters
Objective: Perform spliced alignment of strand-specific RNA-seq data with STAR, generating a BAM file properly tagged for downstream quantification.
Materials & Reagents:
- STAR Aligner (version 2.7.10a or higher).
- Genome Index generated with
--sjdbGTFfileoption. - FASTQ files (paired-end or single-end).
- High-performance computing node (≥ 32 GB RAM for mammalian genomes).
Procedure:
- Run STAR alignment. The key parameter is
--outSAMstrandField intronMotif, which instructs STAR to infer strand information from the intron motif (GT-AG) and tag each read in the BAM file appropriately. - Output Files:
sample_star_Aligned.sortedByCoord.out.bam: Main genomic alignments for visualization and quantification.sample_star_ReadsPerGene.out.tab: Raw gene-level counts. Caution: Ensure the strandedness option (columns 2-4) is correctly selected in downstream analysis.sample_star_Aligned.toTranscriptome.out.bam: Transcriptomic alignments for tools like Salmon.
Visualized Workflows
Diagram 1: Stranded Library Analysis Workflow
Stranded Library Analysis Workflow
Diagram 2: Strand Orientation in Common Protocols
Read Orientation in Stranded Protocols
The Scientist's Toolkit
Table 3: Essential Research Reagents and Tools
| Item | Function/Description | Example Product/Software |
|---|---|---|
| Stranded mRNA Library Prep Kit | Incorporates dUTPs during second strand synthesis, enabling strand preservation. | Illumina TruSeq Stranded mRNA Kit |
| Stranded Total RNA Library Prep Kit | Depletes rRNA and preserves strand information, ideal for degraded or FFPE samples. | Illumina TruSeq Stranded Total RNA Kit |
| STAR Aligner | Spliced aligner capable of detecting splice junctions and tagging alignments with strand info. | STAR (open source) |
| RSeQC Toolkit | Suite of tools for RNA-seq quality control, includes infer_experiment.py. |
RSeQC Python package |
| featureCounts | Fast and accurate read quantification program that accepts strand-specific parameters. | Part of Subread package |
| Salmon / kallisto | Alignment-free quantification tools that model library type for accurate transcript-level estimates. | Salmon (select -l ISR for dUTP) |
| High-Quality Reference Annotation | Comprehensive, non-redundant gene annotation (GTF/BED) is essential for strandedness inference. | GENCODE, RefSeq |
| SAMtools | Manipulates and indexes alignment files (BAM/SAM) for downstream analysis. | SAMtools (open source) |
Best Practices for Reproducibility and Scalability in High-Throughput Pipelines
Within the context of a thesis on performing read alignment with STAR for RNA-seq research, establishing reproducible and scalable high-throughput pipelines is critical. This protocol details best practices to ensure that STAR alignment workflows produce consistent, verifiable results that can be efficiently scaled from single samples to large cohorts, a necessity for robust scientific discovery and drug development.
Application Note 1: Foundational Principles for Reproducible Pipelines
Reproducibility requires capturing the exact computational environment and parameters used. For STAR alignment, this extends beyond the core command.
Key Data and Software Versioning Table
| Component | Recommended Practice | Example/Tool |
|---|---|---|
| STAR Version | Pin exact version; avoid "latest". | STAR 2.7.11a |
| Reference Genome | Document source, version, and build. | GENCODE Human Release 43 (GRCh38.p13) |
| Annotation File | Use version-matched GTF/GFF3. | gencode.v43.annotation.gtf |
| Read Files | Use immutable, checksum-verified files. | FASTQ with MD5 sums |
| Containerization | Package entire software environment. | Docker/Singularity image |
| Workflow Manager | Define pipeline steps explicitly. | Nextflow, Snakemake, WDL |
| Parameter File | Store all non-default parameters in a config file. | star_alignment_config.yaml |
| Random Seed | Set a fixed seed for any stochastic steps. | --outSAMseed 2024 |
Protocol 1.1: Generating a Reproducible STAR Index
This protocol must be executed once per genome/annotation combination and the output archived.
Materials & Reagents: High-memory compute node (≥32GB RAM for mammalian genome), STAR software, reference genome FASTA, annotation GTF.
Methodology:
- Data Acquisition: Download and verify checksums for the reference genome FASTA and annotation GTF from a trusted source (e.g., GENCODE, Ensembl).
- Environment Capture: Create a Dockerfile or Singularity definition file specifying the OS and exact STAR version.
- Index Generation Command:
- Archive: Store the final index directory with a README detailing all inputs and the exact command used.
Diagram 1: Reproducible STAR Index Generation Workflow
Application Note 2: Scalable Alignment Execution
Scalability ensures efficient processing of hundreds to thousands of samples without manual intervention or error.
Key Performance Metrics Table (Representative Data)
| Metric | Small Scale (10 samples) | Large Scale (1000 samples) | Best Practice for Scaling |
|---|---|---|---|
| Wall Clock Time | ~5 hours | ~500 hours | Parallelize per sample; use cluster/batch. |
| CPU Efficiency | ~85% | Can drop to <60% | Monitor with tools like htop; optimize threads. |
| I/O Bottleneck | Minimal | Severe | Use local SSDs for temp files; optimize storage. |
| Memory/Job | ~32GB | ~32GB | Request consistent memory; use --limitOutSJcollapsed. |
| Failure Rate | 0% | May be 2-5% | Implement automated re-try logic for known errors. |
Protocol 2.1: Implementing a Scalable STAR Alignment Workflow
Using a workflow manager to parallelize alignment across a sample sheet.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Pipeline |
|---|---|
| Sample Manifest (CSV) | Central table linking sample ID to FASTQ paths and metadata. |
| STAR Container Image | Ensures identical software environment across all compute nodes. |
| Configuration File (YAML) | Decouples parameters (genomeDir, threads) from workflow logic. |
| Cluster Scheduler (e.g., SLURM) | Manages resource allocation and job queueing for high-throughput execution. |
| Centralized Storage | High-performance, shared filesystem (e.g., Lustre, NFS) for inputs and outputs. |
| Log Aggregator | Collects and indexes Log.final.out files from all jobs for centralized review. |
Methodology:
- Create Sample Manifest:
samples.csvwith columns:sample_id, read1_path, read2_path. - Define Workflow (e.g., Nextflow):
- Execute with Scaling:
nextflow run main.nf -profile cluster,slurm -resume
Diagram 2: Scalable RNA-seq Alignment Pipeline Architecture
Application Note 3: Validation and Reporting
Ensuring pipeline outputs are consistent and correct across runs.
Protocol 3.1: Post-Alignment Quality Aggregation
Create a consolidated report from all samples for rapid assessment.
Methodology:
- Extract Key Metrics: Write a script (e.g., in Python) to parse
*Log.final.outfiles for metrics:- Uniquely mapped reads %
- Multi-mapped reads %
- Reads mapped to too many loci %
- Mismatch rate per base
- Generate Summary Table: Aggregate into a single CSV file.
- Create Visualization: Use R or Python to plot cohort-level statistics (e.g., distribution of mapping rates).
- Compare Runs: If re-running, use statistical tests (e.g., correlation) to compare key metrics between pipeline executions to detect silent failures or regressions.
Diagram 3: Validation and Quality Control Workflow
Validating STAR Results and Comparing Aligners for Your Research Goals
Post-alignment quality control (QC) is a critical step in RNA-seq analysis to ensure data integrity and the reliability of downstream results. This guide, part of a broader thesis on RNA-seq alignment with STAR, details key metrics, tools, and protocols for validating your aligned BAM files.
Critical Post-Alignment QC Metrics
A successful STAR alignment must be assessed using multiple quantitative and qualitative metrics. The table below summarizes the core categories and their ideal values.
Table 1: Key Post-Alignment QC Metrics and Interpretations
| Metric Category | Specific Metric | Ideal Value/Range | Interpretation & Implication |
|---|---|---|---|
| Alignment Yield & Efficiency | Overall Alignment Rate | > 70-80% | Percentage of total reads aligned to the genome. Low rates may indicate poor RNA quality or contamination. |
| Uniquely Mapped Reads | > 70% of aligned | Reads mapped to a single genomic location. Crucial for accurate quantification. | |
| Multi-mapped Reads | ~10-20% of aligned | Reads mapped to multiple locations. High percentages are expected in RNA-seq but can complicate analysis. | |
| Mapping Quality | Read Mapping Quality (MAPQ) | High proportion with MAPQ ≥ 255 (STAR's unique mapping score) | Distribution of mapping quality scores. A high peak at 255 indicates confident unique mappings. |
| Read Distribution | Reads in Genes (Exonic) | > 60-70% of aligned | Proportion of reads falling within annotated exonic regions. Low values may indicate high intronic/ intergenic reads from genomic DNA contamination. |
| Reads in Introns | < 20-30% | High levels can indicate incomplete RNA enrichment or high nascent RNA. | |
| Reads in Intergenic Regions | < 10-15% | High levels often signal genomic DNA contamination. | |
| Strandedness | Reads Assigned to Sense/ Antisense Strand | Matches library prep (e.g., ~99% sense for stranded dUTP) | Verifies the correctness of the strandedness parameter used in alignment and quantification. |
| Coverage Uniformity | 5'->3' Coverage Bias | Even across transcript body | Significant bias indicates degradation (3' bias) or library prep issues. |
| Insert Size | Median Insert Size | Matches expected fragment length (~200-300bp for standard prep) | Validates the library construction and alignment performance. |
Essential QC Tools: Qualimap and RSeQC
Two comprehensive tools are widely used for calculating the metrics in Table 1.
Qualimap (rna-seq mode)
A user-friendly tool that generates an HTML report with extensive plots and statistics from a BAM file and GTF annotation.
Protocol 1: Running Qualimap for RNA-seq QC
- Prerequisite: Install Qualimap (e.g., via conda:
conda install -c bioconda qualimap). Ensure you have a BAM file from STAR and the corresponding GTF annotation file. - Basic Command:
- Output: Navigate to the output directory and open the
qualimapReport.htmlfile in a web browser. Key sections include "Genomic Origin of Reads" (exonic/intronic/intergenic pie chart), "Reads Genomic Position" (coverage bias plot), and alignment statistics.
RSeQC
A suite of Python scripts offering granular control over specific QC modules, ideal for batch processing and scripting.
Protocol 2: Running Key RSeQC Modules
- Prerequisite: Install RSeQC (e.g.,
pip install RSeQCorconda install -c bioconda rseqc). - Execute Core Modules:
- Read Distribution:
read_distribution.py -i /path/to/your_alignment.bam -r /path/to/hg38_RefSeq.bed - Inner Distance (Insert Size):
inner_distance.py -i /path/to/your_alignment.bam -o /path/to/output_prefix -r /path/to/hg38_RefSeq.bed - Gene Body Coverage:
geneBody_coverage.py -i /path/to/your_alignment.bam -o /path/to/output_prefix -r /path/to/hg38_RefSeq.bed - Strand Specificity:
infer_experiment.py -i /path/to/your_alignment.bam -r /path/to/hg38_RefSeq.bed
- Read Distribution:
- Output Analysis: Each module generates text files and/or PNG plots. The
read_distribution.pyoutput should be compared to Table 1.infer_experiment.pyoutputs the empirically determined strandedness.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for RNA-seq Alignment & Validation
| Item | Function in STAR Alignment/Validation |
|---|---|
| High-Quality Total RNA | Starting material. RIN > 8 is typically recommended for most applications to ensure intact mRNA. |
| Strand-Specific Library Prep Kit (e.g., Illumina TruSeq Stranded mRNA) | Creates cDNA libraries where the original strand information is preserved, essential for accurate transcript quantification. |
| STAR Aligner Software | Spliced aligner specifically designed for RNA-seq data. Performs fast and accurate mapping to the reference genome. |
| Reference Genome FASTA & Annotation GTF | The genomic sequence and gene model annotations for the organism of study. Must be from the same source (e.g., both from Ensembl). |
| Qualimap or RSeQC Software | Post-alignment QC toolkits that calculate critical metrics and generate visual reports from BAM files. |
| Computational Resources (High RAM Server/Cluster) | STAR alignment and indexing are memory-intensive (> 32GB RAM for mammalian genomes). |
Visualization of the Post-Alignment QC Workflow
Post-Alignment Quality Control Decision Workflow
Integrated Experimental Protocol for Alignment Validation
Protocol 3: Comprehensive STAR Alignment Validation Pipeline This protocol assumes STAR alignment has been completed.
- Generate Alignment Statistics: Use
samtools statson the BAM file to get basic alignment metrics (total reads, alignment rate). - Run Qualimap for Holistic Assessment: Follow Protocol 1. Examine the HTML report, focusing on the "Genomic Origin" pie chart and "Coverage across genes" plot for uniformity.
- Run RSeQC for Specific Metrics: Follow Protocol 2. Use
infer_experiment.pyto confirm strandedness andread_distribution.pyto get exact percentages for exonic/intronic/intergenic reads. - Cross-Check and Interpret: Compare metrics from all sources against the benchmarks in Table 1.
- Action on Low Exonic Rate: Check for DNA contamination (review Bioanalyzer/Fragment Analyzer traces of the RNA and final library). Consider using a ribosomal RNA depletion kit if not already used.
- Action on High 3' Bias: Indicates RNA degradation. Check RNA Integrity Number (RIN) from the initial sample; proceed with caution as quantification may be biased towards 3' ends of transcripts.
- Action on Low Alignment Rate: Verify the reference genome matches the species. Check for adapter contamination in raw reads using FastQC/MultiQC.
- Document and Proceed: Only proceed to quantification (e.g., with featureCounts or RSEM) if all key metrics are within acceptable ranges. Document all QC results for publication.
Within the broader thesis on performing read alignment with STAR for RNA-seq research, it is critical to understand the landscape of modern RNA-seq analysis tools. STAR is a fundamental, splice-aware aligner that maps reads to a reference genome. This application note places STAR in context by comparing it to the aligner HISAT2 and the pseudo-aligners/quasi-mappers Salmon and Kallisto, which quantify transcript abundance directly without full genome alignment. The choice of tool dictates downstream analysis, balancing accuracy, resource needs, and biological question.
Comparative Analysis of Tools
Table 1: Core Algorithmic & Functional Comparison
| Feature | STAR | HISAT2 | Salmon | Kallisto |
|---|---|---|---|---|
| Primary Method | Spliced alignment to genome | Hierarchical FM-index to genome | Pseudo-alignment/lightweight mapping to transcriptome | Pseudo-alignment via de Bruijn graph of k-mers |
| Reference Needed | Genome + annotations | Genome + annotations | Transcriptome (or genome with decoys) | Transcriptome |
| Key Output | Aligned SAM/BAM files (splice-aware) | Aligned SAM/BAM files (splice-aware) | Transcript-level counts & TPM | Transcript-level counts & TPM |
| Handles Splicing | Yes, explicitly via annotations | Yes, via global & local FM-indexes | Implicitly, via provided transcriptome | Implicitly, via provided transcriptome |
| Speed | Very Fast mapping, high memory | Fast, lower memory than STAR | Extremely Fast quantification | Extremely Fast quantification |
| Memory Usage | High (~30GB+ for human) | Moderate (~10GB for human) | Low | Very Low |
| Quantification Accuracy | Good, used with counting tools (e.g., featureCounts) | Good, used with counting tools | High, models sequence & GC bias | High, with bootstraps for uncertainty |
Table 2: Typical Performance Metrics (Human RNA-seq, ~30M paired-end reads)*
| Metric | STAR | HISAT2 | Salmon (selective alignment) | Kallisto |
|---|---|---|---|---|
| Wall-clock Time | 30-60 minutes | 60-90 minutes | 15-25 minutes | 10-20 minutes |
| Peak Memory (GB) | 28-32 GB | 8-12 GB | 4-6 GB | < 4 GB |
| Alignable Read % | 85-90%+ | 85-90%+ | N/A (direct quantification) | N/A (direct quantification) |
| Typical Use Case | Novel splice junction discovery, variant analysis, detailed genomic visualization | Standard splicing analysis on limited resources | High-throughput, accurate transcript quantification | Ultra-fast, standard transcript quantification |
*Data synthesized from current benchmark literature (2023-2024). Actual performance depends on hardware, read length, and parameters.
Experimental Protocols
Protocol 1: Basic RNA-seq Alignment Workflow with STAR
Application: Generate aligned BAM files for downstream variant, splice junction, or counting analysis.
- Generate Genome Index: (One-time step)
STAR --runThreadN [threads] --runMode genomeGenerate --genomeDir /path/to/GenomeDir --genomeFastaFiles genome.fa --sjdbGTFfile annotation.gtf --sjdbOverhang [read_length - 1] - Align Reads:
STAR --genomeDir /path/to/GenomeDir --readFilesIn sample_R1.fastq.gz sample_R2.fastq.gz --readFilesCommand zcat --runThreadN [threads] --outSAMtype BAM SortedByCoordinate --outFileNamePrefix sample_aligned. - Output:
sample_aligned.Aligned.sortedByCoord.out.bamis ready for downstream tools.
Protocol 2: Transcript Quantification with Salmon (Alignment-based Mode)
Application: Accurate, bias-aware transcript-level quantification leveraging genome alignment for mapping.
- Generate Decoy-aware Salmon Index:
Use
salmon indexwith a transcriptome fasta and a decoy file (list of chromosome names) to reduce spurious mappings. - Quantify using Selective Alignment (via STAR):
First, align with STAR using
--quantMode TranscriptomeSAMto output alignments translated to transcript coordinates. Then, run Salmon:salmon quant -t transcriptome.fa -l A -a sample_aligned.toTranscriptome.out.bam -o salmon_quant --gencode
Protocol 3: Direct Quantification with Kallisto
Application: Fast, single-step transcript abundance estimation.
- Build Kallisto Index:
kallisto index -i transcriptome.idx transcriptome.fasta - Run Quantification:
kallisto quant -i transcriptome.idx -o output_dir -t [threads] --bias sample_R1.fastq.gz sample_R2.fastq.gz
Visualizations
Title: RNA-seq Analysis Tool Decision Workflow
Title: Key Steps in STAR's Spliced Alignment Algorithm
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Software for RNA-seq Alignment Analysis
| Item | Function & Relevance |
|---|---|
| High-Quality Reference Genome & Annotation (GTF/GFF) | Foundational files for alignment (STAR, HISAT2) and transcriptome definition (Salmon, Kallisto). Ensembl or GENCODE sources are standard. |
| Computational Resource (High RAM Node) | Critical for STAR. Requires server/node with >32GB RAM for human genome alignment. HISAT2 and pseudo-aligners can run on standard desktops. |
| FASTQ Quality Control Tool (FastQC/MultiQC) | Assess read quality, adapter contamination, and GC content before alignment, ensuring input data integrity for all tools. |
| Alignment Post-processing Tools (SAMtools, Picard) | Manipulate, sort, index, and assess quality of BAM files output by STAR/HISAT2 (e.g., samtools flagstat). |
| Quantification Aggregation Tool (tximport/tximeta) | Essential downstream step to import Salmon/Kallisto transcript-level estimates into R/Bioconductor for gene-level differential expression analysis. |
| Differential Expression Suite (DESeq2, edgeR) | Ultimate goal of many pipelines. These packages require count matrices derived from aligned BAMs (via featureCounts) or imported from pseudo-aligners. |
Application Notes
High-quality read alignment with STAR is a critical prerequisite for robust downstream RNA-seq analysis. The choice of alignment parameters directly influences gene- and isoform-level quantifications, which form the foundational count matrix for differential expression (DE) tools like DESeq2 and edgeR. Inaccurate alignment, particularly at splice junctions, introduces quantification bias that propagates through the analysis pipeline, compromising the statistical detection of differentially expressed genes and isoforms.
DESeq2 and edgeR, while using different statistical frameworks (negative binomial generalized linear models), are both sensitive to the quality of input counts. STAR's alignment precision affects:
- Read Count Accuracy: Misalignments lead to incorrect assignment of reads to genes, skewing expression estimates.
- Multimapping Resolution: STAR's handling of reads mapping to multiple loci influences the uniqueness of counts, a critical factor for DE sensitivity.
- Splice Junction Discovery: Comprehensive junction detection by STAR is essential for isoform quantification tools (e.g., Salmon, Cufflinks), which in turn affect differential isoform usage (DIU) analysis.
Table 1: Impact of STAR Alignment Parameters on Downstream DE Analysis
| STAR Parameter | Primary Function | Impact on DESeq2/edgeR Input | Recommended Setting for DE |
|---|---|---|---|
--outFilterMultimapNmax |
Max number of loci a read can map to | Higher values increase non-unique counts, adding noise. | 1 (for simple genomes) or 10 (with --winAnchorMultimapNmax) |
--outSAMprimaryFlag |
Defines primary alignment criteria | Ensures a single, best count per read for the gene count matrix. | AllBestScore (default) |
--outFilterMismatchNmax |
Max number of mismatches per read pair | Too high reduces precision; too low decreases alignment rate. | Scale with read length (e.g., 10 for 2x100bp) |
--quantMode |
Enables direct transcript/gene quantification | --quantMode GeneCounts provides a ready-made count table. |
GeneCounts and/or TranscriptomeSAM |
--twopassMode |
Two-pass alignment for novel junction discovery | Increases sensitivity for novel isoforms, improving isoform-level analysis. | Basic for novel isoform discovery |
Table 2: Comparison of DESeq2 and edgeR in Context of STAR-Generated Counts
| Feature | DESeq2 | edgeR |
|---|---|---|
| Core Model | Negative Binomial GLM with shrinkage estimators. | Negative Binomial GLM with empirical Bayes methods. |
| Dispersion Estimation | Moderates dispersion estimates towards a trended mean. | Estimates a common, trended, and tagwise dispersion. |
| Handling of Low Counts | More robust via automatic independent filtering. | Can be sensitive; requires pre-filtering of lowly expressed genes. |
| Isoform Analysis | Not native; requires isoform counts from tools like tximport. | Not native; requires isoform counts from external quantification. |
| Key Strength | Stability with small sample sizes and complex designs. | Speed and flexibility for large datasets. |
Protocols
Protocol 1: STAR Alignment for Gene-Level Differential Expression Analysis
Objective: Generate a gene-level count matrix from paired-end RNA-seq reads suitable for DESeq2/edgeR.
- Generate Genome Index:
STAR --runMode genomeGenerate --genomeDir /path/to/genomeDir --genomeFastaFiles GRCh38.primary_assembly.fa --sjdbGTFfile gencode.v44.annotation.gtf --sjdbOverhang 99 - Align Reads:
STAR --genomeDir /path/to/genomeDir --readFilesIn sample_R1.fastq.gz sample_R2.fastq.gz --readFilesCommand zcat --runThreadN 12 --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 10 --winAnchorMultimapNmax 50 --outSAMprimaryFlag AllBestScore --quantMode GeneCounts - Collate Counts: The gene count matrix is in the
*ReadsPerGene.out.tabfiles from all samples. Column 2 (unstranded) or 3/4 (stranded) is used. - Import into DESeq2 (R):
Protocol 2: STAR Alignment for Isoform Quantification and Differential Usage
Objective: Generate alignments suitable for isoform quantification with Salmon (pseudoalignment) or StringTie.
- Align to Genome with Transcriptome Output:
STAR --genomeDir /path/to/genomeDir --readFilesIn sample_R1.fastq.gz sample_R2.fastq.gz --readFilesCommand zcat --runThreadN 12 --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM --outFilterMultimapNmax 10 - Convert to Fastq for Salmon: Use samtools to extract reads aligned to the transcriptome from the
Aligned.toTranscriptome.out.bamfile:samtools fastq Aligned.toTranscriptome.out.bam > sample_transcriptome.fastq - Run Salmon (in mapping-based mode):
salmon quant -t GRCh38.transcripts.fa -a sample_transcriptome.fastq -o salmon_quant --validateMappings - Import Isoform Counts into DESeq2 via tximport (R):
Diagrams
Title: STAR Alignment Pathways to Differential Expression Analysis
Title: DESeq2 and edgeR Core Analysis Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for STAR Alignment and Downstream DE Analysis
| Item | Function & Relevance |
|---|---|
| High-Quality Reference Genome & Annotation (GTF/GFF3) | Essential for STAR genome indexing. Ensures accurate splice junction mapping and gene/transcript identification. Use Ensembl or GENCODE for model organisms. |
| STAR Aligner (v2.7.10a+) | Spliced aligner of choice for RNA-seq. Its speed and accuracy in junction mapping directly determine quantification quality. |
| DESeq2 (R/Bioconductor) | Statistical software for differential gene expression analysis. Robust to varying library sizes and effective at handling small sample numbers. |
| edgeR (R/Bioconductor) | Statistical software for differential expression. Efficient and powerful for a wide range of experimental designs, often faster on large datasets. |
| tximport / tximeta (R/Bioconductor) | Critical bridging tools. Import transcript-level abundance estimates (e.g., from Salmon) into DESeq2/edgeR, correcting for potential changes in isoform length. |
| Salmon or kallisto | Pseudoalignment/lightweight quantification tools. Enable accurate transcript-level quantification, often used with STAR's --quantMode TranscriptomeSAM output. |
| SAMtools & BEDTools | For processing and manipulating alignment (BAM) files, including sorting, indexing, and coverage analysis. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | STAR alignment and genome indexing are memory- and CPU-intensive. A server with >32GB RAM and multiple cores is typically required. |
Within the broader thesis on performing RNA-seq read alignment with STAR, this case study addresses the extension of the core STAR algorithm to two complex, modern data types: single-cell RNA sequencing (scRNA-seq) and long-read sequencing (e.g., from PacBio or Oxford Nanopore). While standard STAR is optimized for bulk, short-read RNA-seq, the STARlong mode enables the alignment of long, error-prone reads. This application note details the necessary modifications to experimental design, command-line parameters, and downstream interpretation to successfully align these complex datasets, maintaining the principle of accurate splice junction discovery central to the STAR methodology.
Key Parameter Adjustments for Complex Data
Aligning complex data requires significant deviation from default STAR parameters. The following tables summarize critical adjustments.
Table 1: Core STARlong Parameters for Error-Prone Long Reads
| Parameter | Typical Value for Long Reads | Function & Rationale |
|---|---|---|
--runMode alignReads |
Default | Specifies alignment mode. |
--runThreadN |
[User-defined] | Number of threads. |
--genomeDir |
[Path] | Directory of the STAR-generated genome index. |
--readFilesIn |
[File] | Input FASTQ file(s). |
--readNameSeparator |
space |
Often needed for long-read basecaller output. |
--seedSearchStartLmax |
30 |
Crucial: Increases the initial search seed length to improve accuracy on noisy reads. |
--seedPerReadNmax |
200000 |
Dramatically increases allowed seeds per read to compensate for high error rates. |
--seedPerWindowNmax |
200 |
Increases seeds per window to maintain sensitivity. |
--alignTranscriptsPerReadNmax |
200000 |
Allows mapping of many potential transcriptomes per read. |
--alignEndsType |
Local |
Enables soft-clipping of ends, ideal for adapter sequences or low-quality read termini common in long reads. |
--outSAMtype |
BAM SortedByCoordinate |
Standard sorted BAM output. |
--outFilterMultimapScoreRange |
3 |
Relaxes multi-map score range to retain more alignments. |
--winAnchorMultimapNmax |
200 |
Increases anchors for multi-mapping. |
--outFilterMismatchNmax |
1000 |
Crucial: Allows a very high number of mismatches to accommodate high error rates. |
--scoreGapNoncan |
-20 |
Less severe penalty for non-canonical splice junctions. |
--scoreGapGCAG |
-20 |
Less severe penalty for GCAG splice junctions. |
--scoreGapATAC |
-20 |
Less severe penalty for ATAC splice junctions. |
Table 2: STAR Parameters for Single-Cell (scRNA-seq) Data
| Parameter | Typical Value for scRNA-seq | Function & Rationale |
|---|---|---|
| Standard Alignment Parameters | As per bulk RNA-seq | Initial read alignment uses standard settings. |
--outFilterType |
BySJout |
Reduces over-filtering of reads supporting novel junctions, important for heterogeneous cell populations. |
--limitOutSJcollapsed |
2000000 |
Increases allowed collapsed splice junctions output. |
--alignSJoverhangMin |
8 |
Ensures reliable splice junction detection with shorter overhangs sometimes seen in fragmented scRNA-seq libraries. |
--quantMode |
GeneCounts |
Critical: Outputs read counts per gene directly into ReadsPerGene.out.tab for downstream analysis with tools like Seurat. |
--sjdbGTFfile |
[GTF File] | Essential for accurate gene-level quantification against an annotation. |
| Post-Alignment Processing | [BAM File] | Use featureCounts (from Subread package) or similar for additional granularity if --quantMode GeneCounts is insufficient. |
Detailed Experimental Protocols
Protocol 3.1: Genome Index Generation for scRNA-seq and Long-Read Alignment
- Objective: Generate a STAR genome index optimized for sensitivity.
- Materials: Reference genome FASTA file, annotation GTF file, high-memory compute server.
- Procedure:
- Download the appropriate reference genome (e.g., GRCh38) and corresponding annotation GTF from Ensembl or GENCODE.
- Use the following STAR command to generate the index. This is common for both scRNA-seq and long-read alignment, though
--sjdbOverhangshould be adjusted for read length.
Protocol 3.2: Alignment of PacBio/Nanopore cDNA Reads using STARlong
- Objective: Map error-prone long-read RNA-seq data to a reference genome.
- Materials: Processed long-read FASTQ files, STAR genome index, STARlong-compatible STAR version (>=2.7.0).
- Procedure:
- Pre-process reads (optional but recommended): Perform adapter trimming and quality filtering using tools like
cutadaptorfiltlong. - Execute the STARlong alignment using parameters from Table 1.
- The primary output,
Aligned.sortedByCoord.out.bam, is ready for downstream analysis with tools likeIsoQuantorSQANTI3for isoform identification and quantification.
- Pre-process reads (optional but recommended): Perform adapter trimming and quality filtering using tools like
Protocol 3.3: Alignment of 10x Genomics scRNA-seq Data with STAR
- Objective: Generate a gene expression count matrix from scRNA-seq FASTQ files.
- Materials: Paired-end scRNA-seq FASTQ files (Read1: cell barcode + UMI; Read2: transcript), STAR genome index.
- Procedure:
- Use
--quantMode GeneCountsduring alignment to obtain raw gene counts. STAR correctly assigns reads to genes based on the provided GTF. - The key output file
ReadsPerGene.out.tabcontains raw counts per gene (column 2 for unstranded libraries). This file, alongside barcode whitelisting and UMI deduplication (performed separately by tools likeCell Ranger,starsolo, orUMI-tools), forms the basis for the cell-by-gene count matrix.
- Use
Visualizations
STARlong Workflow for Isoform Discovery
scRNA-seq Alignment to Count Matrix Pipeline
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions & Materials
| Item | Function & Application |
|---|---|
| STAR Aligner (v2.7.10a+) | Core splice-aware alignment software. STARlong functionality is integrated into recent versions. |
| High-Quality Reference Genome & Annotation (GENCODE/Ensembl) | Essential for accurate mapping and gene/isoform quantification. Must match organism and strain. |
| 10x Genomics Cell Ranger | Integrated pipeline for processing 10x scRNA-seq data; uses STAR internally and handles barcode/UMI processing. |
| SAMtools/BEDTools | Universal utilities for processing, indexing, and manipulating SAM/BAM alignment files. |
| IsoQuant or SQANTI3 | Specialized tools for downstream analysis of long-read alignments to characterize isoform diversity and quality. |
| Seurat or Scanpy | Primary R/Python packages for downstream analysis of the cell-by-gene count matrix from scRNA-seq. |
| High-Performance Computing (HPC) Cluster | Essential resource for genome indexing and aligning large-scale sequencing datasets efficiently. |
| Oxford Nanopore MinION/GridION or PacBio Sequel IIe | Platforms generating long-read sequencing data for isoform-level analysis. |
Within the broader thesis on performing read alignment with STAR for RNA-seq research, a critical precursor step is selecting the appropriate analytical tool. This decision is not arbitrary but must be driven by the experimental design and the specific biological questions posed. Incorrect tool selection can lead to inaccurate alignments, biased quantification, and erroneous biological conclusions. This Application Note provides a framework for this decision-making process, with a focus on upstream considerations that inform the use of STAR.
Decision Framework for Alignment Tool Selection
The choice of a read aligner like STAR must be evaluated against alternatives based on key project parameters. The following table summarizes primary quantitative and qualitative factors for comparison.
Table 1: Quantitative Comparison of RNA-seq Alignment Tools
| Tool | Algorithm Type | Speed (Relative) | Memory Usage | Splice-Aware | Best Suited For |
|---|---|---|---|---|---|
| STAR | Seed-and-vote, Spliced | Very Fast | High (~30GB for Human) | Yes | Standard RNA-seq, Novel splice discovery |
| HISAT2 | FM-index, Spliced | Fast | Moderate | Yes | Standard RNA-seq, Low-memory environments |
| Kallisto | Pseudoalignment | Very Fast | Low | Yes (via de Bruijn graph) | Transcript quantification only |
| Salmon | Pseudoalignment | Very Fast | Low | Yes (via quasi-mapping) | Rapid transcript quantification |
| Bowtie2 | FM-index, Unspliced | Fast | Low | No | Aligning to cDNA/transcriptome only |
Table 2: Decision Framework Based on Biological Question
| Primary Biological Question | Recommended Tool Class | Key Rationale | STAR Applicability |
|---|---|---|---|
| Novel isoform or splice variant discovery | Spliced, reference-guided aligner (e.g., STAR) | Maximizes sensitivity for unannotated splice junctions. | High. STAR's two-pass mode is ideal for novel junction discovery. |
| Differential gene expression | Pseudoaligner (Kallisto/Salmon) or Spliced Aligner | Speed and accuracy for quantification are paramount. | Medium. Use if genomic-level analysis (e.g., allele-specific expression) is also needed. |
| Viral integration or fusion gene detection | Spliced, reference-guided aligner (e.g., STAR) | Requires precise mapping to genome, often with chimeric read detection. | High. STAR's built-in chimeric detection is a key feature. |
| Small RNA or miRNA profiling | Unspliced, short-read aligner (Bowtie) | Reads are short and map to mature sequences; splicing is irrelevant. | Low. Not optimized for very short reads. |
| Single-cell RNA-seq (droplet-based) | Spliced aligner optimized for speed (STAR, HISAT2) | Must process billions of reads efficiently; UMI-aware pipelines exist. | High. STARsolo is a dedicated, optimized mode for scRNA-seq. |
Detailed Protocol: Two-Pass STAR Alignment for Novel Junction Discovery
This protocol is recommended when the biological question involves discovering unannotated splice junctions or isoforms, a common scenario in cancer or developmental biology studies.
Materials: Research Reagent Solutions
- Computational Server: Minimum 16 cores, 32 GB RAM (64+ GB recommended for mammalian genomes).
- Reference Genome FASTA File: Downloaded from ENSEMBL or UCSC (e.g.,
GRCh38.primary_assembly.genome.fa). - Annotation GTF File: Corresponding to the genome build (e.g.,
gencode.v44.annotation.gtf). - RNA-seq Reads: In FASTQ format (e.g.,
sample_R1.fastq.gz,sample_R2.fastq.gz). - STAR Software: Version 2.7.10a or later.
- SAMtools: For processing alignment outputs.
Method
Part 1: Genome Index Generation
- Prepare Directory and Files: Place the genome FASTA and annotation GTF files in a dedicated directory. Create an output directory for the index.
- Generate Genome Index: Run STAR in
--runMode genomeGenerate. Critical parameters include--sjdbOverhang, which should be read length minus 1 (e.g., 99 for 100bp paired-end reads).
Part 2: First Pass Alignment
- Align Reads: For each sample, perform alignment, directing novel splice junction information to a file.
The key output is
sample_firstPass_SJ.out.tab.
Part 3: Second Pass with Novel Junctions
- Merge Junctions: Combine
SJ.out.tabfiles from all samples if processing a multi-sample experiment. - Re-generate Genome Index with Novel Junctions: Incorporate the merged junction file into a new genome index.
- Final Alignment: Align reads using the new index to produce the final BAM file.
The final, sorted BAM file is
sample_final_Aligned.sortedByCoord.out.bam, and gene-level counts are insample_final_ReadsPerGene.out.tab.
Visualizing the Decision and Experimental Workflow
Decision and Workflow for RNA-seq Alignment
Two-Pass Logic for Sensitivity
Conclusion
Mastering STAR for RNA-seq alignment is a fundamental skill that bridges raw sequencing data and biologically meaningful insights. This guide has walked through the foundational logic of spliced alignment, provided a robust methodological pipeline, offered solutions for common pitfalls, and emphasized the importance of validation. A well-executed STAR alignment ensures the integrity of all subsequent analyses, from differential expression in disease models to novel isoform detection in cancer research. As RNA-seq applications expand into clinical diagnostics and personalized medicine, incorporating best practices for alignment—including the emerging use of population-specific genomes and long-read integration—will be crucial for generating reliable, actionable genomic data that can accelerate drug discovery and improve patient outcomes.