Unlocking Immune Activation: The Critical Role of H12 Helix Dynamics in NBS-LRR Proteins
This article provides a comprehensive analysis of the activation mechanisms of Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) proteins, focusing on the pivotal conformational dynamics of the H12 (Helix 12) motif.
Unlocking Immune Activation: The Critical Role of H12 Helix Dynamics in NBS-LRR Proteins
Abstract
This article provides a comprehensive analysis of the activation mechanisms of Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) proteins, focusing on the pivotal conformational dynamics of the H12 (Helix 12) motif. We explore the foundational structural biology of the NBS domain, detailing how H12 acts as a molecular switch for immune signaling. Methodological approaches for studying these dynamics, including computational simulations and advanced biophysical assays, are reviewed. Common experimental challenges and optimization strategies for capturing these transient states are addressed. Finally, we compare H12 dynamics across different NBS-LRR subfamilies and validate its role through mutational studies and disease-linked variants. This synthesis aims to inform researchers and drug developers targeting this crucial node in plant and mammalian immunity.
The Molecular Switch: Understanding H12 Helix Dynamics in NBS-LRR Activation
Nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins constitute a primary class of intracellular immune receptors in plants, responsible for detecting pathogen effectors and initiating robust defense responses. This whitepaper provides an in-depth technical guide on their structure, function, and activation mechanism, with a specific focus on the dynamics of the H12 helix within the NB-ARC domain—a critical regulatory element for transitioning from autoinhibited to active states.
Structural Architecture and Classification
NBS-LRR proteins are modular receptors typically composed of three core domains:
- N-terminal Domain: Often a Toll/Interleukin-1 receptor (TIR) or Coiled-coil (CC) domain involved in downstream signaling.
- Central NB-ARC Domain: A nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4, functioning as a molecular switch regulated by ADP/ATP binding and hydrolysis.
- C-terminal LRR Domain: Acts primarily as a sensor domain for direct or indirect effector recognition.
Table 1: Major NBS-LRR Subclasses and Features
| Subclass | N-terminal Domain | Key Structural Motifs | Example Proteins | Activation Model |
|---|---|---|---|---|
| TNL | TIR | TIR, NB-ARC, LRR | RPS4, SNC1 | Direct effector binding or guardee surveillance. Often require EDS1 helper protein. |
| CNL | Coiled-coil (CC) | CC, NB-ARC, LRR | RPS5, MLA10 | Direct effector binding or decoy integration. Often require NRG1 helper protein. |
| RNL | RPW8-like CC | CC(NB-ARC), LRR | ADR1, NRG1 | Often function as helper NBS-LRRs amplifying signals from sensor TNLs/CNLs. |
Activation Mechanism and H12 Helix Dynamics
The NB-ARC domain is the regulatory core, with conformational changes governed by nucleotide state. The H12 helix (also known as the MHD motif-containing helix) is a critical latch stabilizing the autoinhibited state.
Pre-activation State: The receptor is primed with ADP, and the H12 helix is engaged with the NB subdomain, locking the LRR domain in a closed conformation. Effector Recognition: Direct or indirect effector binding to the LRR domain disrupts its interaction with the NB-ARC domain. Nucleotide Exchange & H12 Release: The loss of LRR inhibition promotes ADP-to-ATP exchange. This induces a major conformational shift in the NB-ARC, particularly the release and rotation of the H12 helix. Active State: The H12 helix disengages, allowing the N-terminal domain to oligomerize and form a signaling-competent resistosome, which recruits downstream signaling components to execute hypersensitive response (HR) and Systemic Acquired Resistance (SAR).
Diagram 1: NBS-LRR Activation via H12 Helix Release
Key Experimental Protocols for Studying H12 Dynamics
Site-Directed Mutagenesis of the MHD Motif
Purpose: To probe the functional role of the H12 helix by altering key residues. Protocol:
- Primer Design: Design complementary oligonucleotide primers containing the desired point mutation (e.g., altering the conserved Asp residue in the MHD motif).
- PCR Amplification: Perform high-fidelity PCR using a plasmid containing the wild-type NBS-LRR gene as template.
- DpnI Digestion: Treat the PCR product with DpnI endonuclease (targets methylated parental DNA) to eliminate template plasmids.
- Transformation: Transform the digested product into competent E. coli cells for cloning.
- Validation: Isolate plasmid DNA and confirm the mutation by Sanger sequencing.
In Vitro Nucleotide Binding & Hydrolysis Assays
Purpose: To quantify the impact of H12 mutations on ATPase activity. Protocol:
- Protein Purification: Express and purify recombinant NB-ARC domain (wild-type and H12 mutant) with an affinity tag (e.g., GST, His6).
- ATPase Assay Setup: In a 96-well plate, mix 2 µg of protein with reaction buffer (20 mM Tris-HCl pH 7.5, 5 mM MgCl2) and 1 mM ATP. Include a no-protein control.
- Incubation & Detection: Incubate at 25°C for 60 minutes. Use a commercial colorimetric phosphate assay kit (e.g., Malachite Green) to measure inorganic phosphate (Pi) release.
- Data Analysis: Calculate hydrolysis rates from a phosphate standard curve. Compare mutant vs. wild-type activity.
Table 2: Quantitative ATPase Activity of H12 Mutants
| Protein Variant | Conserved Motif | ATP Hydrolysis Rate (nmol Pi/min/µg) | Relative Activity (%) | Proposed Effect |
|---|---|---|---|---|
| Wild-type NB-ARC | Intact MHD | 8.7 ± 0.5 | 100% | Basal, controlled hydrolysis |
| H12 Mutant (D->V) | MHV | 0.9 ± 0.2 | 10% | Loss of hydrolysis, often autoactive |
| H12 Mutant (H->A) | MAD | 12.3 ± 1.1 | 141% | Enhanced hydrolysis, often inactive |
Co-Immunoprecipitation (Co-IP) for Interaction Studies
Purpose: To assess how H12 mutations affect intramolecular domain interactions or resistosome assembly. Protocol:
- Plant Infiltration: Transiently co-express in Nicotiana benthamiana leaves: (a) an NBS-LRR (WT or mutant) tagged with GFP, and (b) a potential interactor (e.g., another NBS-LRR domain) tagged with Myc.
- Protein Extraction: At 48 hours post-infiltration, homogenize leaf tissue in non-denaturing extraction buffer with protease inhibitors.
- Immunoprecipitation: Incubate lysates with anti-GFP nanobeads for 2 hours at 4°C. Wash beads thoroughly.
- Immunoblotting: Elute proteins and separate by SDS-PAGE. Probe membranes with anti-GFP and anti-Myc antibodies to detect pulled-down complexes.
Diagram 2: Co-IP Workflow for NBS-LRR Interactions
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for NBS-LRR/H12 Helix Research
| Reagent / Material | Function & Application | Key Considerations |
|---|---|---|
| pEAQ-HT Expression Vector | High-yield transient protein expression in plants via agroinfiltration. | Ideal for expressing full-length NBS-LRRs, which are often difficult in prokaryotic systems. |
| Maltose-Binding Protein (MBP) Fusion Tag | Enhances solubility of recombinant NB-ARC domains for in vitro assays. | MBP can be cleaved off post-purification to avoid interference with conformational studies. |
| Anti-Phospho-p44/42 MAPK Antibody | Detects activation of MAPK cascades, a rapid downstream readout of NBS-LRR activation. | Validates functional output of receptor signaling in plant tissue. |
| Non-hydrolyzable ATP Analogs (ATPγS, AMP-PNP) | Used to trap the NB-ARC domain in a specific nucleotide-bound state for structural studies. | Critical for X-ray crystallography or Cryo-EM of active-state complexes. |
| Nicotiana benthamiana EDS1 Knockout Line | Genetic background for functional assays of TNL proteins, which require EDS1. | Essential for dissecting specific helper protein requirements. |
| Microscale Thermophoresis (MST) Instrument | Quantifies binding affinities (Kd) between purified LRR domains and pathogen effectors. | Requires fluorescently labeled protein; uses very small sample volumes. |
Architecture of the Nucleotide-Binding Site (NBS) Domain
This whitepaper details the architecture of the nucleotide-binding site (NBS) domain, a core component of nucleotide-binding, leucine-rich-repeat (NLR) immune receptors in plants and animals. Its structure and conformational dynamics are fundamental to understanding immune signaling activation. The content is framed within a broader thesis investigating the role of the H12 helix (part of the winged-helix domain, WHD) dynamics in the transition of NBS-LRR proteins from autoinhibited to active states. Precise molecular architecture dictates the ADP/ATP-binding status and subsequent oligomerization, forming signaling-active inflammasomes or resistosomes.
Core Architectural Features of the NBS Domain
The NBS domain is a member of the signal transduction ATPases with numerous domains (STAND) family. Its conserved architecture consists of a central nucleotide-binding pocket flanked by several subdomains that regulate its activity.
Key Subdomains:
- Nucleotide-Binding Pocket (NB-ARC): Comprises the Walker A (P-loop), Walker B, and Sensor 1 motifs that coordinate the phosphate groups of ADP/ATP. The Mg²⁺ ion is chelated by residues in the Walker B and Sensor 1 motifs.
- Winged-Helix Domain (WHD): Contains the H12 helix (also referred to as the "MHD2" or "H12 helix" in plant NLRs), which acts as a molecular latch. In the ADP-bound state, the H12 helix packs against the nucleotide-binding pocket, stabilizing autoinhibition.
- Helical Domain 1 (HD1): A bundle of α-helices that interacts with the WHD and LRR domain.
- ARC Subdomain (ARC1/ARC2): Composed of helical repeats that connect the NB to the WHD; critical for transmitting conformational changes.
Quantitative Structural Parameters: Table 1: Key Quantitative Parameters of the NBS Domain Architecture
| Parameter | ADP-Bound (Inactive) State | ATP-Bound (Active) State | Measurement Method |
|---|---|---|---|
| Distance: Walker A to H12 | ~10-15 Å (Close) | ~20-25 Å (Increased) | X-ray Crystallography |
| Rotation of WHD | 0° (Reference) | ~15-25° (Clockwise) | Cryo-EM / Molecular Dynamics |
| H12 Helix Displacement | Packed against NB pocket | Released/Displaced | Hydrogen-Deuterium Exchange MS |
| Nucleotide Occupancy | >95% ADP | >90% ATP | Isothermal Titration Calorimetry |
| Interdomain Salt Bridges | 4-6 stable bridges (e.g., H12 to NB) | 1-2 bridges broken | Computational Simulation |
Detailed Experimental Protocols
Understanding NBS architecture requires multidisciplinary approaches. Below are detailed protocols for key experiments.
Protocol 1: Determining Nucleotide-Binding Affinity via Isothermal Titration Calorimetry (ITC)
- Protein Purification: Express and purify recombinant NBS domain (e.g., residues 1-250 of an NLR) using affinity (Ni-NTA) and size-exclusion chromatography (Superdex 200).
- Sample Preparation: Dialyze protein (~50 µM) into ITC buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl₂). Prepare nucleotide (ADP or ATP) solution in the identical dialysis buffer.
- ITC Run: Load the protein solution into the sample cell (1.4 mL). Fill the syringe with nucleotide solution (typically 0.5-1 mM). Set parameters: 25°C, reference power 10 µcal/s, stirring speed 750 rpm.
- Titration: Perform 19 injections of 2 µL each, with 150-second intervals. A control titration of nucleotide into buffer is mandatory.
- Data Analysis: Subtract control data from sample data. Fit the integrated heat peaks to a single-site binding model using the instrument's software (e.g., MicroCal PEAQ-ITC) to derive stoichiometry (N), binding constant (Kd), enthalpy (ΔH), and entropy (ΔS).
Protocol 2: Probing H12 Helix Dynamics via Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
- Labeling: Dilute purified NBS domain protein (in ADP- or ATP-bound state) 10-fold into D₂O-based labeling buffer (pD 7.5, 25°C). Incubate for varying time points (e.g., 10s, 1min, 10min, 1h).
- Quenching: At each time point, mix labeling reaction 1:1 with quench buffer (0.1 M phosphate, pH 2.2, 0 °C) to reduce pH and temperature, slowing exchange.
- Digestion & Separation: Immediately inject quenched sample into a cooled LC system with an immobilized pepsin column for rapid digestion (<1 min).
- Mass Spectrometry: Trap peptides on a C18 trap column, separate via a C18 analytical column, and analyze with a high-resolution mass spectrometer (e.g., Q-TOF).
- Data Processing: Use specialized software (e.g., HDExaminer) to identify peptides and calculate deuterium incorporation for each time point. Regions like the H12 helix will show increased deuterium uptake (indicating increased solvent exposure/dynamics) upon ATP binding and activation.
Protocol 3: Structural Validation via Site-Directed Mutagenesis & In Vitro ATPase Assay
- Mutagenesis: Design primers to introduce point mutations in key motifs (e.g., Walker A K->A, Sensor 1 S->A, H12 helix conserved residues). Use PCR-based site-directed mutagenesis kit.
- Protein Expression/Purification: Express and purify mutant proteins as in Protocol 1.
- ATPase Activity Assay (Malachite Green): a. Incubate 5 µM protein in reaction buffer (20 mM HEPES pH 7.5, 5 mM MgCl₂, 1 mM DTT) with 1 mM ATP for 30 min at 25°C. b. Stop reaction by adding equal volume of Malachite Green reagent (0.081% malachite green, 2.32% polyvinyl alcohol, 5.72% ammonium molybdate in 6N HCl). c. Incubate 1 min at room temperature, measure absorbance at 620 nm. d. Calculate liberated phosphate using a standard curve (0-100 µM KH₂PO₄). Walker A mutants should show >90% reduction in activity.
Signaling Pathway and Workflow Visualizations
NBS Activation via H12 Release Pathway
HDX-MS Workflow for Dynamics Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for NBS Domain Architecture Studies
| Reagent / Material | Supplier Examples | Function in Research |
|---|---|---|
| Recombinant NBS Protein | In-house expression; cDNA from Addgene | The core substrate for structural, biochemical, and biophysical analysis. Often tagged (His, GST) for purification. |
| Non-hydrolyzable ATP Analog (ATPγS, AMP-PNP) | Sigma-Aldrich, Jena Bioscience | Traps the NBS domain in a stable "ATP-bound" conformation for structural studies (X-ray, Cryo-EM). |
| HDX-MS Kit & Software | Waters, Thermo Fisher, HDExaminer | Provides standardized buffers and analysis pipelines for reproducible hydrogen-deuterium exchange experiments. |
| Size-Exclusion Chromatography Columns (Superdex 200) | Cytiva | Critical for purifying monodisperse, properly folded NBS protein and analyzing its oligomeric state. |
| Malachite Green Phosphate Assay Kit | Sigma-Aldrich, Cayman Chemical | Quantifies ATP hydrolysis activity to assess the functional impact of architectural mutations (e.g., in H12). |
| Cryo-EM Grids (Quantifoil R1.2/1.3) | Quantifoil, EMS | Used for plunge-freezing NBS domain oligomers (e.g., activated complexes) for high-resolution structure determination. |
| Site-Directed Mutagenesis Kit (Q5) | New England Biolabs | Enables precise alanine scanning or functional knock-in mutations in the NBS, WHD, and H12 helix motifs. |
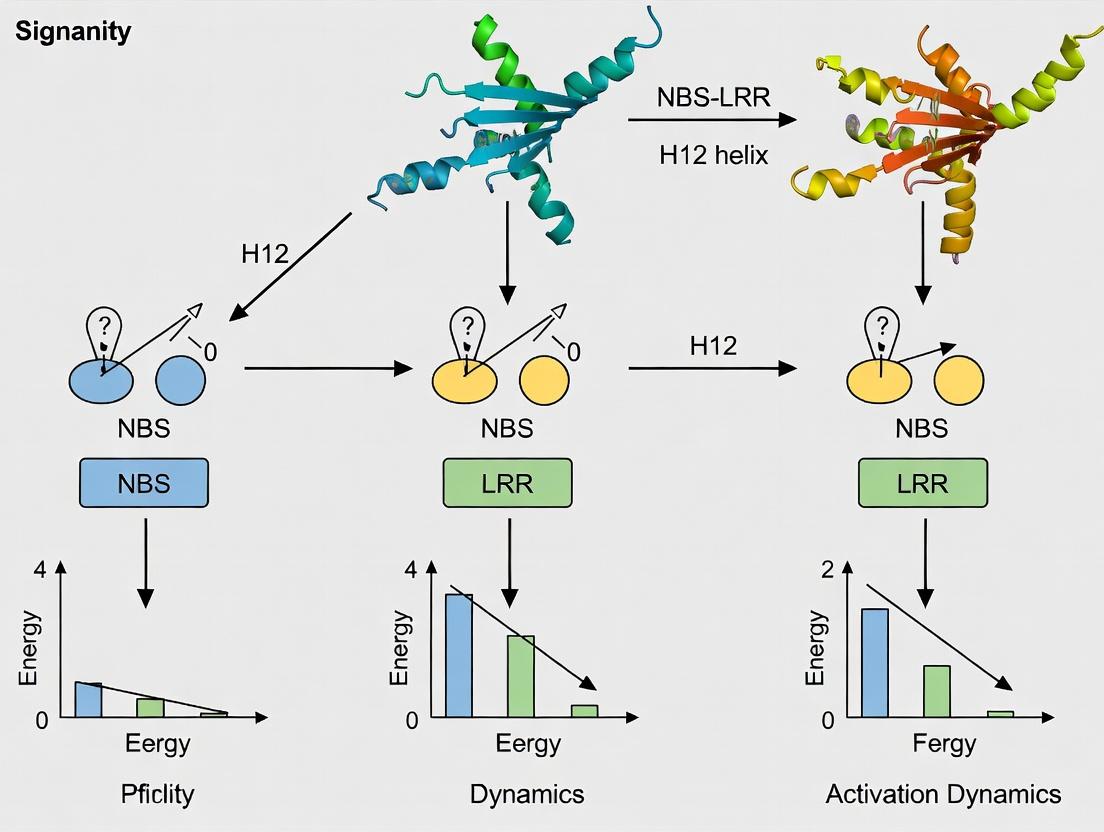
1. Introduction and Thesis Context
Within the broader thesis on Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) protein dynamics and activation, the H12 helix (also frequently termed the "HD2 helix" or "final ARC2 subdomain helix") emerges as a critical structural linchpin. This in-depth technical guide defines the H12 helix through its sequence signatures, three-dimensional architecture, and evolutionarily conserved motifs, framing it as a central regulatory module. The transition of NBS-LRR proteins from autoinhibited to active states is governed by conformational changes propagated through domains like the ARC2, with the H12 helix serving as a pivotal sensor and transducer. Precise definition of H12 is therefore fundamental for research aimed at elucidating immune signaling mechanisms and for drug development targeting NBS-LRR pathologies.
2. Sequence Definition and Conserved Motifs
The H12 helix is located at the C-terminal end of the ARC2 (Apaf-1, R proteins, and CED-4) subdomain within the NB-ARC (Nucleotide-Binding adaptor shared by APAF-1, R proteins, and CED-4) domain. Its sequence spans approximately 20-30 residues and is characterized by a pattern of hydrophobic and charged amino acids that dictate its structure and function.
Table 1: Conserved Sequence Motifs within the H12 Helix Region
| Motif Name | Consensus Sequence | Position Relative to H12 | Functional Role |
|---|---|---|---|
| MHD | Met-His-Asp/Asn | Immediately C-terminal to H12 | Critical for nucleotide binding/hydrolysis; the His is essential for autoinhibition. |
| RNBS-D | (F/L)La(F/Y)xxLxL | Forms the core of the H12 helix | Stabilizes the helix via hydrophobic packing; "xx" often charged (K/R, D/E) for dynamics. |
| Walker B | DDLD/E | N-terminal to H12 (precedes it) | Coordinates Mg²⁺ ion for ATP hydrolysis; mutation disrupts signaling. |
The MHD motif is not part of the helical structure itself but is a definitive landmark for its C-terminal boundary. The integrity of the RNBS-D motif is essential for maintaining the autoinhibited state.
3. Structural Definition and Dynamics
In the autoinhibited, ADP-bound state, the H12 helix packs tightly against the NB subdomain and the rest of the ARC2, forming part of a compact hydrophobic core that sequesters the MHD histidine. This packing prevents spontaneous activation.
Table 2: Structural Parameters of H12 Helix in Auto-Inhibited vs. Active States
| Parameter | ADP-Bound (Inactive) State | ATP-Bound/Transition State | Method of Determination |
|---|---|---|---|
| Orientation | Packed parallel to ARC1/NB interface | Unwound/Displaced | X-ray Crystallography, MD Simulations |
| Key Interactions | Hydrophobic packing with RNBS-A, H-bond from MHD-His to ADP β-phosphate | Disrupted; MHD-Asp may engage new partners | Mutagenesis & ITC |
| B-Factor/Dynamics | Low B-factor (stable) | Increased B-factor / high mobility | Cryo-EM, Crystallography |
| Role of MHD His | Coordinated, essential for lock | Displaced, breaking the "lock" | Functional Studies |
Upon pathogen effector perception (often via LRR domain rotation), nucleotide exchange (ADP to ATP) occurs. This induces a major conformational shift in the NB-ARC, where the H12 helix, along with the entire ARC2 subdomain, undergoes a ~130° rotation and translational shift. This "domain swing" releases the MHD motif, exposing interfaces for downstream signaling partners like the N-terminal domains (TIR or CC).
Title: H12 Helix Role in NBS-LRR Activation Pathway
4. Experimental Protocols for H12 Characterization
4.1. Site-Directed Mutagenesis of H12/MHD Motifs
- Objective: To probe the functional role of specific residues.
- Protocol:
- Design primers incorporating the desired point mutation (e.g., MHD His->Ala).
- Perform PCR using a plasmid containing the wild-type NBS-LRR gene as a template with a high-fidelity polymerase.
- Digest the parental (methylated) template DNA with DpnI.
- Transform the nuclease-treated product into competent E. coli, plate on selective agar, and screen colonies by sequencing.
- Clone the mutated fragment back into the full-length expression vector.
4.2. In Vitro ATPase Activity Assay
- Objective: To quantify the effect of H12 mutations on nucleotide hydrolysis.
- Protocol:
- Express and purify recombinant NB-ARC or full-length protein (wild-type and H12 mutants).
- In a reaction buffer (e.g., 20 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl₂), mix protein (1-5 µM) with [γ-³²P]ATP or a fluorescent ATP analog.
- Incubate at 25°C. Withdraw aliquots at time points (0, 5, 15, 30, 60 min).
- Stop the reaction with EDTA. Spot aliquots on a Polyethylenimine-cellulose TLC plate.
- Develop the TLC plate in 0.5 M LiCl / 1 M formic acid. Visualize and quantify the ratio of ATP to ADP using a phosphorimager. Calculate hydrolysis rates.
4.3. Differential Scanning Fluorimetry (Thermal Shift)
- Objective: To assess the structural stability of H12 mutants and ligand binding.
- Protocol:
- Prepare protein samples (0.2 mg/mL) in a compatible buffer with a fluorescent dye (e.g., SYPRO Orange).
- Pipette samples into a 96-well PCR plate. For binding studies, add ligands (ADP, ATPγS, dATP) at varying concentrations.
- Use a real-time PCR instrument to ramp the temperature from 25°C to 95°C at a rate of ~1°C/min, monitoring fluorescence.
- Plot fluorescence vs. temperature. Determine the melting temperature (Tm) as the inflection point. A shift in Tm indicates altered stability or ligand binding.
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for H12 Helix and NBS-LRR Dynamics Research
| Reagent/Material | Function/Application | Example/Note |
|---|---|---|
| Site-Directed Mutagenesis Kits | Introduces precise mutations in H12/MHD motifs for functional studies. | Q5 Site-Directed Mutagenesis Kit (NEB), QuickChange. |
| Bac-to-Bac Baculovirus System | Expression of full-length, post-translationally modified large NBS-LRR proteins for structural biology. | Invitrogen Bac-to-Bac system for insect cell expression. |
| Non-hydrolyzable ATP Analogs | Traps the protein in an activation-competent state for structural analysis. | ATPγS, AMP-PNP, ADP·BeF₃. |
| Size-Exclusion Chromatography (SEC) Columns | Assesses oligomeric state (monomer vs. active oligomer) of wild-type vs. H12 mutants. | Superose 6 Increase, Superdex 200 (Cytiva). |
| Anti-FLAG/Strep-Tactin Beads | For co-immunoprecipitation assays to test H12 mutant effects on downstream partner binding. | Immunoprecipitation of tagged NBS-LRR protein complexes. |
| Microscale Thermophoresis (MST) Kit | Quantifies binding affinities (Kd) between purified H12 peptide variants and nucleotides/partners. | Monolith NT.115 instrument and capillaries. |
| Cryo-EM Grids & Vitrobot | Prepares samples for high-resolution structural determination of full-length activated complexes. | Quantifoil R1.2/1.3 gold grids, Thermo Fisher Vitrobot. |
6. Conclusion
The H12 helix is definitively characterized as a conserved structural element within the NB-ARC domain whose conformation is directly governed by nucleotide state. Its sequence motifs, particularly the RNBS-D and the adjacent MHD, form a functional unit that acts as a nucleotide-sensitive switch. Within the thesis of NBS-LRR activation dynamics, the H12 helix is the linchpin of the domain swing mechanism, translating nucleotide exchange into large-scale structural reorganization. Precise experimental definition of its properties is thus critical for understanding immune signaling and for designing interventions that modulate this switch, offering targets for drug development in inflammatory and autoimmune diseases.
Within the broader thesis on NBS-LRR (Nucleotide-Binding Site Leucine-Rich Repeat) protein dynamics, the activation mechanism pivots on conformational changes in the NB-ARC (Nucleotide-Binding Adaptor Shared by APAF-1, R proteins, and CED-4) domain. This whitepaper investigates the "ATP Lid" hypothesis, which posits that the H12 helix (part of the MHD motif) functions as a critical gatekeeper, sterically regulating access to the nucleotide-binding pocket. Its precise positioning, governed by the ADP/ATP binding state, dictates the transition between inactive ("off") and active ("on") signaling states, a fundamental process in plant immunity and inflammatory pathways.
Mechanistic Basis of the Hypothesis
The NB-ARC domain cycles between ADP-bound (inactive) and ATP-bound (active) states. The "ATP Lid," encompassing the H12 helix and adjacent loops, undergoes a pronounced conformational shift.
- ADP-Bound State (Off): H12 is positioned over the nucleotide-binding pocket, creating a closed conformation that stabilizes ADP binding and prevents spontaneous activation.
- ATP-Bound State (On): Upon pathogen effector perception, ADP/ATP exchange occurs. The binding of ATP, with its extra phosphate, induces a steric clash and electrostatic repulsion. This forces the H12 helix to swing away from the pocket, forming an open conformation. This displacement unmasks key interaction surfaces (e.g., the EDVID motif) necessary for oligomerization and downstream signaling cascade initiation.
Table 1: Structural Metrics of H12 in Different Nucleotide States
| Metric | ADP-Bound State (Inactive) | ATP-Bound State (Active) | Measurement Technique |
|---|---|---|---|
| H12 Displacement | 0 Å (Reference) | 8-12 Å | X-ray Crystallography |
| Rotation Angle | 0° (Reference) | 30-45° | Molecular Dynamics |
| Solvent Accessibility of Binding Pocket | Low (< 15%) | High (> 60%) | Computational Analysis |
| K_d for Nucleotide | ~0.1 µM (ADP) | ~2.0 µM (ATP) | Isothermal Titration Calorimetry |
Table 2: Functional Impact of H12 Mutations in Model NBS-LRR Proteins
| Protein (Organism) | H12 Mutation | Observed Phenotype | Signaling Output (Relative to WT) |
|---|---|---|---|
| MLA10 (Barley) | D478V (MHD) | Autoactive | Constitutive (150-200%) |
| NOD2 (Human) | R444C (Near H12) | Loss-of-function | Abolished (<5%) |
| APAF-1 (Human) | K160E (H12 Stabilizer) | Non-functional | Abolished (<2%) |
| NRC4 (Plant) | G427E (H12 Pivot) | Enhanced sensitivity | Hyperactive (250%) |
Key Experimental Protocols
Site-Directed Mutagenesis & Functional Complementation
Purpose: To validate the role of specific H12 residues in gating. Protocol:
- Design primers incorporating the desired point mutation (e.g., converting Asp to Val in the MHD motif).
- Perform PCR amplification on the wild-type NBS-LRR cDNA clone using a high-fidelity polymerase.
- Digest the parental methylated DNA with DpnI.
- Transform the mutated plasmid into competent E. coli, screen colonies, and sequence-validate.
- Transfect mutated constructs into an appropriate cell line (e.g., HEK293T for NLRs, plant protoplasts for R proteins).
- Measure signaling output via reporter assays (NF-κB luciferase for human NLRs, HR cell death assays for plants).
Limited Proteolysis Coupled to Mass Spectrometry
Purpose: To probe nucleotide-dependent conformational changes in the ATP lid. Protocol:
- Purify recombinant NB-ARC domain protein.
- Incubate separate samples with 1 mM ADP or the non-hydrolyzable ATP analog AMP-PNP.
- Subject each sample to limited digestion with a broad-specificity protease (e.g., subtilisin) for varying time intervals (0-30 min).
- Quench reactions with protease inhibitors and analyze by SDS-PAGE for differential banding patterns.
- For MS analysis, digest samples with trypsin and analyze via LC-MS/MS to identify protected or exposed regions, specifically monitoring H12-derived peptides.
Hydrogen-Deuterium Exchange (HDX) Mass Spectrometry
Purpose: To map dynamic changes in solvent accessibility of the ATP lid region. Protocol:
- Prepare protein samples in ADP- and ATP-bound states.
- Dilute samples into D₂O-based exchange buffer and allow deuterium incorporation for defined time points (10s to 4 hours).
- Quench exchange at low pH and 0°C.
- Rapidly digest with immobilized pepsin, inject onto a UPLC-MS system maintained at 0°C.
- Measure mass increase of peptide fragments. Decreased deuterium uptake in H12 indicates stabilization/closing; increased uptake indicates dynamic opening.
Visualizations
H12 Gating in NBS-LRR Activation
HDX-MS Protocol for H12 Dynamics
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Investigating the ATP Lid
| Reagent / Material | Function & Application in H12 Research |
|---|---|
| Non-hydrolyzable ATP Analogs (AMP-PNP, ATPγS) | Used to lock the NB-ARC domain in an ATP-bound state for structural studies (X-ray, Cryo-EM) without hydrolysis. |
| Site-Directed Mutagenesis Kits (e.g., Q5) | For introducing precise point mutations into the H12 helix (MHD motif) to study gain/loss-of-function phenotypes. |
| Nucleotide-Agarose Beads (ADP/ATP) | Affinity purification of NB-ARC domain proteins and assessment of nucleotide-binding affinity in pull-down assays. |
| Hydrogen-Deuterium Exchange (HDX) MS Buffer Kits | Standardized buffers and quench solutions for reproducible measurement of H12 conformational dynamics. |
| Conformation-Specific Antibodies | Antibodies raised against peptides mimicking the "open" or "closed" H12 state to detect active/inactive populations in cells. |
| Thermal Shift Dye (e.g., SYPRO Orange) | Used in differential scanning fluorimetry to measure stabilization/destabilization of the domain by H12 mutations. |
| Recombinant NLR/NBS-LRR Proteins | High-purity, full-length or NB-ARC domain proteins for in vitro biochemical and biophysical assays. |
The nucleotide-binding switch is a universal molecular mechanism governing protein activation. Within the NBS-LRR (Nucleotide-Binding Site Leucine-Rich Repeat) family of plant immune receptors and their NLR (NOD-Like Receptor) homologs in mammals, this switch dictates the transition from an autoinhibited standby state to an active signaling oligomer. This whitepaper delves into the atomic-level conformational journey, with a specific focus on the pivotal role of the H12 helix (also known as the HD1/hWH loop) dynamics. The broader thesis posits that the displacement and structural reorganization of the H12 helix, triggered by ATP binding and hydrolysis at the NBS, are the central drivers of the allosteric changes that culminate in the formation of the active resistosome/inflammasome complex. Understanding this precise trajectory is critical for designing agonists or antagonists that can modulate immune responses for therapeutic or agricultural purposes.
Structural & Energetic States of the NBS Domain
The NBS domain exhibits distinct conformational and energetic states dependent on its bound nucleotide.
Table 1: Quantitative Characterization of NBS States
| State Parameter | ADP-Bound (Inactive) | Nucleotide-Free (Apo) | ATP-Bound (Active) | Experimental Method (Typical) |
|---|---|---|---|---|
| H12 Helix Position | Packed against core, blocking oligomerization interface | Disordered/ flexible | Displaced/ Reoriented, exposing interface | X-ray Crystallography, HDX-MS |
| NBS Domain Conformation | Closed, compact | Open, unstable | Closed, tense | SAXS, Cryo-EM |
| Solvent Accessibility of H12 | Low (~10-15%) | High (~40-60%) | Intermediate (~20-30%) | HDX-MS, ASA Calculation |
| Kd for Nucleotide | N/A (tightly bound) | Low affinity (µM-mM range) | High affinity (nM-µM range) | ITC, SPR |
| Free Energy (ΔG) of State | Lowest (most stable) | Highest (least stable) | Intermediate, energy stored | Computational MD Simulation |
| Hydrolysis Rate (kcat) | Very Slow | N/A | Fast (activated) | Malachite Green Phosphate Assay |
Detailed Experimental Protocols for Key Analyses
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) for H12 Dynamics
Objective: To measure the relative solvent accessibility and dynamics of the H12 helix region in different nucleotide states.
Protocol:
- Sample Preparation: Purify recombinant NBS-LRR protein (e.g., Arabidopsis ZAR1). Prepare three conditions: (i) with 2mM ADP, (ii) apo (treated with apyrase), (iii) with 2mM ATPγS (non-hydrolyzable analog).
- Deuterium Labeling: Dilute protein 10-fold into D₂O-based labeling buffer (20mM HEPES pD 7.5, 150mM NaCl). Incubate at 25°C for five time points (e.g., 10s, 1min, 10min, 1h, 4h).
- Quenching & Digestion: Quench by adding equal volume of pre-chilled quench buffer (400mM KH₂PO₄/H₃PO₄, pH 2.2, 2M Guanidine HCl). Immediately pass over an immobilized pepsin column at 0°C.
- LC-MS/MS Analysis: Desalt peptides on a C18 trap column and separate with a C18 UPLC column (gradient 5-35% acetonitrile in 0.1% formic acid over 12 min). Analyze with a high-resolution mass spectrometer (e.g., Q-TOF).
- Data Processing: Use dedicated software (e.g., HDExaminer) to identify peptides and calculate deuterium uptake. The H12-derived peptide will show decreased uptake in ADP state (protected) and increased uptake in apo/ATP states (more exposed/dynamic).
Surface Plasmon Resonance (SPR) for Nucleotide Binding Kinetics
Objective: To determine the binding affinity (Kd), on-rate (ka), and off-rate (kd) of ATP/ADP to the NBS domain.
Protocol:
- Sensor Chip Functionalization: Use a nitrilotriacetic acid (NTA) chip. Inject 0.5 mM NiCl₂ for 2 min to charge the surface.
- Ligand Immobilization: Inject His-tagged NBS domain (10 µg/mL in running buffer: 20mM HEPES pH 7.5, 150mM NaCl, 50µM EDTA) for 4-5 min to achieve ~5000 RU response.
- Analyte Binding: Inject a series of nucleotide concentrations (ATPγS or ADP from 0.1 µM to 100 µM) in running buffer at a flow rate of 30 µL/min for 120s association, followed by 300s dissociation.
- Regeneration: Regenerate the surface with two 30s pulses of 350mM EDTA.
- Data Analysis: Double-reference the sensorgrams (buffer blank & zero analyte). Fit the data to a 1:1 Langmuir binding model using the SPR evaluation software to extract ka, kd, and calculate Kd (Kd = kd/ka).
Visualizing the Conformational Pathway
Diagram Title: NBS-LRR Activation Conformational Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for NBS-LRR Conformational Studies
| Reagent / Material | Function & Application | Key Considerations |
|---|---|---|
| ATPγS (Adenosine 5´-[γ-thio]triphosphate) | Non-hydrolyzable ATP analog. Used to trap and stabilize the ATP-bound active conformation for structural studies (X-ray, Cryo-EM). | Prevents signal transduction during assays, allowing isolation of the binding event. |
| Apyrase (Nucleotide Diphosphatase) | Enzymatically removes all nucleotide phosphates (ATP, ADP). Used to generate the nucleotide-free (apo) state of the protein from any starting condition. | Grade and unit activity are critical for complete nucleotide removal without damaging the protein. |
| Deuterium Oxide (D₂O, 99.9%) | The labeling agent for HDX-MS experiments. Allows measurement of hydrogen/deuterium exchange rates to infer protein dynamics and solvent accessibility. | Requires careful handling and storage to prevent back-exchange with atmospheric H₂O. |
| NTA/Ni²⁺ Sensor Chip (e.g., Series S NTA) | For SPR analysis. Provides a reversible, oriented immobilization platform for His-tagged NBS domains to study nucleotide binding kinetics. | Requires nickel charging and careful control of EDTA in buffers to prevent ligand leakage. |
| Size-Exclusion Chromatography (SEC) Column (e.g., Superdex 200 Increase) | Critical for assessing the oligomeric state. Separates monomeric (inactive) from oligomeric (active) complexes following nucleotide incubation. | Must be calibrated with standards. Run in appropriate buffer matching subsequent assays. |
| Cryo-EM Grids (Quantifoil Au R1.2/1.3) | The support film for plunge-freezing protein samples for single-particle cryo-electron microscopy analysis of active oligomers (resistosomes). | Grid quality and freezing conditions are paramount for high-resolution reconstruction. |
Within the broader thesis on NBS-LRR H12 helix dynamics activation research, this whitepaper provides a technical dissection of the critical intermolecular interactions governing the dynamic engagement of the H12 helix with the Leucine-Rich Repeat (LRR) domain. This interface is pivotal for transitioning NBS-LRR immune receptors from an autoinhibited to an active state. We detail the structural, biophysical, and computational methodologies used to probe this interaction, present quantitative data, and outline essential research tools.
Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) proteins are intracellular immune receptors in plants and animals. A conserved mechanistic feature is the autoinhibition of the nucleotide-binding domain (NBD) by intramolecular interactions, primarily involving the H12 helix (also known as the "MHD" or "HD1" motif in some subfamilies) and the LRR domain. Upon pathogen perception, conformational changes disrupt this interface, facilitating ADP/ATP exchange and oligomerization for signal initiation. This guide focuses on experimental approaches to characterize the precise dynamics of H12 disengagement from the LRR and the resulting allosteric signaling.
Table 1: Biophysical Parameters of H12-LRR Interactions
| Parameter | Auto-inhibited State (ADP-bound) | Active State (ATP-bound/Mutant) | Measurement Technique |
|---|---|---|---|
| Buried Surface Area (Ų) | 1050 ± 85 | 220 ± 45 | X-ray Crystallography |
| # of Hydrogen Bonds | 8 - 12 | 1 - 3 | Molecular Dynamics (MD) Simulation |
| ΔG of Binding (kcal/mol) | -9.2 ± 1.1 | -2.1 ± 0.8 | Isothermal Titration Calorimetry (ITC) |
| H12 B-Factor (Ų) Avg. | 45.3 | 78.6 | Cryo-EM / Crystallography |
| Distance H12 Cα to LRR Core (Å) | 4.5 ± 0.7 | 12.8 ± 2.1 | FRET / DEER Spectroscopy |
Table 2: Key Mutational Analysis Impact on Activation
| Mutation Site (H12) | Phenotype | Effect on H12-LRR KD (nM) | Method of Validation |
|---|---|---|---|
| R→A (Salt Bridge) | Constitutive Activation | 8500 (from 120) | ITC, Yeast-two-hybrid |
| D→N (Hydrogen Bond) | Enhanced Auto-inhibition | 65 | SPR, Co-IP |
| Hydrophobic Patch L→A | Loss-of-function | No binding detected | ITC, Structural Analysis |
Core Experimental Protocols
Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS) for Dynamics
Objective: To map solvent accessibility changes in the H12 and LRR regions upon nucleotide exchange. Protocol:
- Sample Preparation: Purify recombinant NBS-LRR protein (e.g., full-length or NBD-LRR construct). Prepare samples in 10 mM HEPES, 150 mM NaCl, pH 7.4.
- Labeling Reaction: Dilute protein to 10 µM in D2O-based buffer containing 5 mM ADP or ATPγS. Incubate at 25°C for 10s, 30s, 1min, 5min, 30min.
- Quenching & Digestion: Quench with pre-chilled 0.5% formic acid (final pH ~2.5). Pass over an immobilized pepsin column (2°C) for 3 min.
- LC-MS/MS Analysis: Desalt peptides on a C18 trap column, separate via RP-UPLC, and analyze with a high-resolution mass spectrometer.
- Data Processing: Use software (e.g., DynamX, HDExaminer) to identify peptides and calculate deuterium uptake differences. Regions protected in ADP-state but exhibiting increased exchange in ATP-state indicate dynamic release.
Double Electron-Electron Resonance (DEER) Spectroscopy
Objective: To measure precise distance distributions between H12 and the LRR domain in solution. Protocol:
- Spin Labeling: Introduce cysteine mutations at strategic sites on H12 (e.g., S451) and the LRR (e.g., T780). Label with MTSSL spin probe.
- Sample Preparation: Purify and concentrate labeled protein to ~100 µM in deuterated buffer with 5% glycerol. Add ADP or ATPγS.
- DEER Measurement: Perform 4-pulse DEER experiments at 50 K on a pulsed EPR spectrometer (e.g., Q-band). Use a standard π/2-τ1-π-τ1-echo-τ2-π-τ2-echo sequence.
- Data Analysis: Process raw data with DeerAnalysis. Background subtract, fit distance distributions using Tikhonov regularization. Compare peaks between nucleotide states.
Molecular Dynamics (MD) Simulation of Disengagement
Objective: To simulate atomic-level trajectories of H12 movement relative to the LRR. Protocol:
- System Setup: Use an auto-inhibited structure (PDB: 5LSE) as the starting model. Solvate in a TIP3P water box with 150 mM NaCl. Neutralize the system.
- Equilibration: Minimize energy. Perform NVT (100 ps) and NPT (1 ns) equilibration at 300 K and 1 bar using position restraints on protein heavy atoms.
- Production Run: Run unrestrained simulations for 500 ns - 1 µs per replicate (3 replicates). Perform parallel simulations with ADP and ATP (modeled via parameterization e.g., with CHARMM36 force field).
- Analysis: Calculate root-mean-square deviation (RMSD) of H12, distances between key residue pairs, hydrogen bond lifetimes, and interaction free energies (MM/PBSA).
Signaling Pathways and Workflows
Title: NBS-LRR Activation Pathway via H12-LRR Disengagement
Title: Multidisciplinary Workflow for H12-LRR Interaction Study
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials
| Item | Function / Application in H12-LRR Research | Example / Note |
|---|---|---|
| Recombinant NBS-LRR Proteins | Full-length or domain constructs for in vitro assays. Critical for ITC, SPR, HDX-MS. | Truncated constructs (NBD-LRR) often necessary for solubility. |
| Non-hydrolyzable ATP Analog (ATPγS) | Mimics active ATP-bound state to trap conformational changes without hydrolysis. | Used in structural studies (Cryo-EM) and HDX-MS comparisons. |
| Site-Directed Mutagenesis Kit | To generate H12 and LRR interface mutants (e.g., alanine scans). | Essential for validating key interaction residues. |
| MTSSL Spin Label | Methanethiosulfonate spin label for site-specific cysteine labeling in DEER spectroscopy. | Requires strategically placed cysteine mutations. |
| Deuterium Oxide (D₂O) | Solvent for HDX-MS experiments to enable measurement of hydrogen exchange rates. | >99.9% isotopic purity required. |
| Anti-Phospho-Ser/Thr Antibodies | To detect phosphorylation events on the NBD or H12 upon activation. | Activation marker in in planta or cell-based assays. |
| Size-Exclusion Chromatography (SEC) Column | To assess oligomeric state changes (monomer vs. oligomer) upon nucleotide exchange. | Superdex 200 Increase commonly used. |
| Molecular Dynamics Software Suite | For simulating H12 dynamics (e.g., GROMACS, AMBER, NAMD). | CHARMM36 or AMBER ff19SB force fields recommended. |
Capturing the Conformational Shift: Techniques to Probe H12 Helix Dynamics
This whitepaper details the application of Molecular Dynamics (MD) simulations to study the flexibility and conformational trajectories of the H12 helix within Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) immune receptors. Understanding H12 dynamics is central to a broader thesis on NBS-LRR activation mechanisms, which propose that the displacement and structural remodeling of the H12 helix (often part of the MHD motif) is a critical switch from autoinhibited to active states, enabling downstream immune signaling. Precise modeling of these dynamics is essential for researchers and drug development professionals aiming to design novel plant disease resistance strategies or small molecule regulators.
Core Computational Methodology
System Preparation
- Initial Structure: Use a high-resolution crystal or cryo-EM structure of an NBS-LRR protein (e.g., ZAR1, MLA10, or mammalian NLRP proteins) in its ADP-bound (inactive) state. The PDB ID 6J5T (ZAR1 resistosome) serves as a key reference for the active state.
- Solvation: Embed the protein in an explicit solvent box (e.g., TIP3P water model) with a minimum 10 Å cushion from the protein to the box edge.
- Neutralization & Ionization: Add counterions (e.g., Na⁺, Cl⁻) to neutralize system charge, followed by additional ions to simulate physiological concentration (e.g., 150 mM NaCl).
- Force Field Parameterization: Apply a modern force field (e.g., CHARMM36m, AMBER ff19SB) suitable for proteins and nucleotides. Special attention is required for parametrizing bound ADP/ATP/dATP.
Simulation Protocol
The following table summarizes a standard multi-step equilibration and production protocol performed using engines like GROMACS, NAMD, or OpenMM.
Table 1: Standard MD Simulation Protocol for NBS-LRR H12 Analysis
| Stage | Description | Duration (ps) | Ensemble | Restraints (Force Constant kJ/mol/nm²) |
|---|---|---|---|---|
| Energy Minimization | Steepest descent to remove steric clashes. | Until convergence (≤1000 steps) | N/A | None |
| NVT Equilibration | Heating to target temperature (310 K). | 100-250 | NVT (Constant Number, Volume, Temperature) | Heavy protein atoms (1000) |
| NPT Equilibration | Pressure coupling to 1 bar. | 100-250 | NPT (Constant Number, Pressure, Temperature) | Protein backbone (400), then (100) |
| Production Run | Unrestrained data collection phase. | 100 ns - 1 µs+ | NPT | None |
- Integration Time Step: 2 fs, with bonds involving hydrogen constrained (e.g., LINCS algorithm).
- Temperature Control: Langevin dynamics or Nosé-Hoover thermostat.
- Pressure Control: Parrinello-Rahman barostat.
- Long-Range Electrostatics: Particle Mesh Ewald (PME) method.
Enhanced Sampling for H12 Transitions
To overcome the timescale limitations of standard MD and capture the full displacement of H12, enhanced sampling techniques are employed:
- Targeted MD (tMD): Application of a steering force to guide the protein from the inactive to the active conformation along a collective variable (CV), such as the root-mean-square deviation (RMSD) of H12.
- Umbrella Sampling (US): A series of simulations (windows) where the position of H12 is restrained at different points along a predefined reaction coordinate (e.g., distance between the center of mass of H12 and the NB-ARC domain). The weighted histogram analysis method (WHAM) is used to reconstruct the free energy landscape.
- Metadynamics: Deposition of repulsive Gaussian potentials in the space of CVs (e.g., H12 dihedral angles, distances) to encourage exploration and compute free energy surfaces.
Key Analyses and Quantitative Metrics
Table 2: Key Quantitative Metrics for Analyzing H12 Dynamics from MD Trajectories
| Analysis Type | Specific Metric | Description & Relevance to H12 Activation |
|---|---|---|
| Conformational Stability | Root Mean Square Deviation (RMSD) of H12 backbone. | Measures global drift of H12; stable low RMSD indicates autoinhibition, large shifts suggest activation. |
| Local Flexibility | Root Mean Square Fluctuation (RMSF) per residue. | Identifies highly flexible residues within H12 and adjacent loops; key hinges for motion. |
| Helical Integrity | Local helix parameters (e.g., helical radius, rise, twist) via helanal or MDAnalysis. |
Quantifies kinking or unwinding of H12 during simulations. |
| Essential Dynamics | Principal Component Analysis (PCA) of Cα positions. | Identifies dominant collective motions; the first 2-3 PCs often describe H12 bending/rotation. |
| Free Energy | ΔG (kcal/mol) along H12 reaction coordinate from US/Metadynamics. | Quantifies the energy barrier for H12 displacement and stability of intermediate states. |
| Intermolecular Contacts | Hydrogen bond occupancy (%) and minimum salt bridge distance (Å). | Tracks breaking of H12-NB-ARC contacts and formation of new H12-LRR or H12-ADP contacts. |
| Solvent Accessibility | Solvent Accessible Surface Area (SASA) of H12 residues (nm²). | Increase in SASA indicates exposure of H12 to solvent upon release from NB-ARC. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Tools for Experimental Validation of H12 Dynamics
| Item | Function in H12 Dynamics Research |
|---|---|
| Wild-type & Mutant NBS-LRR Clones | Site-directed mutagenesis of H12 (e.g., MHD motif mutants) to validate computational predictions on stability and autoinhibition. |
| Non-hydrolyzable Nucleotide Analogs (e.g., AMP-PNP, ADP-BeF₃) | To lock the nucleotide-binding pocket in specific states (ATP-like or ADP-bound) for structural studies and activity assays. |
| Size-Exclusion Chromatography (SEC) Column | To assess oligomeric state (monomer vs. oligomer) of wild-type vs. H12 mutant proteins, linking dynamics to activation. |
| Cross-linking Reagents (e.g., BS³, DSS) | To probe proximity changes between H12 and other domains (NB-ARC, LRR) in different nucleotide states. |
| Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) | To experimentally measure solvent accessibility and dynamics of H12 regions, directly comparable to MD-predicted SASA and fluctuations. |
| Cryo-EM Grids (e.g., Quantifoil Au R1.2/1.3) | For high-resolution structural determination of transient conformations or active oligomers captured via cryo-electron microscopy. |
| Luciferase-based Reporter Assay Kit (for mammalian NLRs) | To quantify the functional consequence of H12 mutations on downstream inflammatory signaling activation in cells. |
| Plant Transfection System (e.g., Agrobacterium, protoplast system) | To perform in planta cell death assays for plant NBS-LRRs with H12 mutations, testing gain/loss-of-function. |
Integrated Pathway & Workflow Visualization
Title: Computational-Experimental Workflow for H12 Dynamics
Title: H12 Dynamics in NBS-LRR Activation Pathway
Site-Directed Spin Labeling (SDSL) and Electron Paramagnetic Resonance (EPR) Spectroscopy
Nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins are critical intracellular immune receptors in plants, responsible for detecting pathogen effectors and initiating defense signaling. A central hypothesis in the field posits that the transition from a resting to an activated state is governed by major conformational changes, particularly within the nucleotide-binding domain. The H12 helix (part of the MHD motif) is theorized to act as a molecular switch. Its dynamics—specifically, its movement away from the nucleotide-binding pocket—is believed to be a key event in activation, facilitating ADP/ATP exchange and subsequent oligomerization. This whitepaper details the application of SDSL-EPR spectroscopy to probe these precise molecular dynamics, providing a technical guide for investigating NBS-LRR activation mechanisms.
Technical Foundations of SDSL-EPR
Principles of Site-Directed Spin Labeling
SDSL involves the selective introduction of a paramagnetic nitroxide radical, typically a methanethiosulfonate (MTSL) derivative, at a specific cysteine residue engineered into a protein of interest. The sensitivity of the nitroxide's EPR spectrum to its local environment and mobility provides a reporter on protein conformation, dynamics, and intermolecular interactions.
EPR Spectroscopic Modalities for Dynamics
- Continuous Wave (CW) EPR: Measures the derivative absorption spectrum. Line shape and spectral breadth report on the rotational correlation time (τc) of the spin label, distinguishing ordered, disordered, or immobilized states.
- Double Electron-Electron Resonance (DEER/PELDOR): Measures dipolar coupling between two spin labels, yielding distance distributions between sites (1.5–8 nm), crucial for detecting conformational changes and oligomerization.
Experimental Protocol for NBS-LRR H12 Dynamics
Sample Preparation
Step 1: Cysteine-Substitution Mutagenesis. Design primers to introduce a single cysteine residue at the desired position within or flanking the H12 helix (e.g., residue D503 in the MHD motif of an archetypal NBS-LRR). Use standard site-directed mutagenesis (e.g., QuikChange) on the plasmid encoding the NBS-LRR protein. Verify by DNA sequencing. Step 2: Protein Expression and Purification. Express the cysteine mutant and a cysteine-less (wild-type background) control protein in an appropriate system (e.g., E. coli or insect cells). Purify via affinity chromatography (e.g., His-tag) under non-reducing conditions. Step 3: Spin Labeling. React the purified protein (50-100 µM) with a 5-10 fold molar excess of (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate (MTSL) for 12-16 hours at 4°C in labeling buffer (e.g., 50 mM Tris, 150 mM NaCl, pH 7.0). Remove excess spin label using a desalting column or extensive dialysis. Step 4: Sample Preparation for EPR. Concentrate spin-labeled protein to ~100-200 µM. For CW-EPR, load 5-10 µL into a glass capillary. For DEER, add 20-25% (v/v) deuterated glycerol as a cryoprotectant and mix with D₂O-based buffer to enhance signal, then load into a quartz EPR tube.
EPR Data Acquisition
CW-EPR Protocol: Acquire spectra at X-band (~9.5 GHz) at a controlled temperature (e.g., 25°C or 10 K). Typical parameters: modulation amplitude 1-2 G, microwave power 5-20 mW, scan range 100 G. DEER Protocol: Perform measurements at Q-band (34 GHz) or X-band at 50 K using a four-pulse DEER sequence: π/2(νobs) – τ1 – π(νobs) – t – π(νpump) – (τ1 + τ2 - t) – π(νobs) – τ2 – echo. The pump frequency is set to the center of the spectrum, and the observer frequency is offset by ~70 MHz. Total acquisition time is typically 8-24 hours.
Data Analysis
CW-EPR: Analyze spectral line shapes. A mobile component yields sharp lines; an immobilized component broadens the spectrum. Spectral simulations yield rotational correlation times. DEER: Process the raw echo decay using DeerAnalysis software. Background subtract to isolate the modulated component, and use Tikhonov regularization to extract the distance distribution profile.
Key Quantitative Data & Findings in NBS-LRR Research
Table 1: Exemplary SDSL-EPR Data from NBS-LRR H12 Helix Studies
| Protein State | Spin Label Position | CW-EPR Mobility (τc, ns) | DEER Distance to Reference Site (Å) | Inferred Conformational State |
|---|---|---|---|---|
| Apo (No Nucleotide) | H12 (503C) | 1.8 ± 0.3 | 45 ± 3 | Dynamic, disordered H12 |
| ADP-Bound (Inactive) | H12 (503C) | 4.5 ± 0.5 | 32 ± 2 | H12 locked over NB pocket |
| ATP-Bound (Active) | H12 (503C) | 2.5 ± 0.4 | 52 ± 4 | H12 released, mobile |
| Activating Mutant (ADP) | H12 (503C) | 2.9 ± 0.4 | 48 ± 3 | H12 partially released |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for SDSL-EPR Studies of NBS-LRR Proteins
| Item | Function / Description | Example Product/Catalog # |
|---|---|---|
| MTSL Spin Label | Thiol-reactive nitroxide radical for cysteine labeling. | (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate (Toronto Research Chemicals, O875000) |
| Cysteine-less Vector | Expression plasmid with all native cysteines mutated, providing a clean background for SDSL. | Custom engineered pET or pFastBac vectors. |
| Deuterated Glycerol | Cryoprotectant for DEER experiments; reduces dielectric loss and extends phase memory time. | Glycerol-d8, 98% (Cambridge Isotope Laboratories, DLM-2775) |
| DEER Analysis Software | Standard package for processing and analyzing DEER data. | DeerAnalysis (www.epr.ethz.ch/software.html) |
| Size-Exclusion Spin Column | For rapid removal of unreacted MTSL post-labeling. | Zeba Spin Desalting Columns, 7K MWCO (Thermo Fisher, 89882) |
Visualization of Methodologies and Pathways
SDSL-EPR Workflow for H12 Dynamics
NBS-LRR Activation Model from EPR Data
SDSL-EPR spectroscopy provides a powerful, residue-level tool for quantifying the conformational dynamics central to NBS-LRR activation. Data robustly support the model of H12 helix displacement as a key mechanistic step. Future work integrating time-resolved EPR with rapid-mix techniques will capture real-time dynamics of activation. Furthermore, applying these methodologies to full-length receptors in native membranes represents the next frontier in understanding plant immune signaling.
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) for Dynamics Mapping
Hydrogen-deuterium exchange mass spectrometry (HDX-MS) has emerged as a powerful biophysical technique for probing the conformational dynamics and solvent accessibility of proteins. When applied within the context of NBS-LRR (Nucleotide-Binding Site Leucine-Rich Repeat) immune receptor activation research, particularly focusing on the H12 helix dynamics, HDX-MS provides unparalleled insights into the allosteric mechanisms underlying plant innate immunity. This whitepaper serves as a technical guide for employing HDX-MS to map the dynamic changes associated with the transition from an auto-inhibited to an active state in NBS-LRR proteins.
Theoretical Foundation and Relevance to NBS-LRR H12 Helix
NBS-LRR proteins are molecular switches. In their resting state, the H12 helix of the NB-ARC domain is proposed to lock the protein via intramolecular interactions. Upon pathogen effector recognition, a major conformational change, potentially involving the displacement or restructuring of the H12 helix, activates downstream signaling. HDX-MS is uniquely suited to monitor these changes by measuring the exchange rate of backbone amide hydrogens for deuteriums from the solvent. Regions that become more solvent-accessible or less structurally ordered upon activation will exhibit increased deuterium uptake, while regions involved in new stabilizing interactions may show decreased uptake.
Core Experimental Protocol for NBS-LRR Dynamics
A generalized, detailed workflow for an HDX-MS experiment targeting NBS-LRR H12 helix dynamics is outlined below.
1. Sample Preparation:
- Protein: Recombinant NBS-LRR protein (e.g., full-length or NB-ARC domain) purified to >95% homogeneity. Two states are required: Apo (auto-inhibited) and Activated (e.g., +ATPγS/Mg²⁺, +effector protein, or oligomerized).
- Buffers: Protein must be in a non-deuterated, low-salt, volatile buffer (e.g., 20 mM HEPES, 100 mM NaCl, pH 7.5). The deuterated buffer must be prepared identically in D₂O (pD read = pH meter read + 0.4).
2. Deuterium Labeling:
- The labeling reaction is initiated by a 10- or 15-fold dilution of the protein solution into the D₂O buffer.
- Reactions are performed at a constant temperature (e.g., 25°C) for multiple time points (e.g., 10 s, 1 min, 10 min, 1 h, 4 h) to capture exchange kinetics.
- A control (t=0) is performed by diluting into H₂O buffer.
- The reaction is quenched at each time point by lowering the pH to 2.5-2.7 (final concentration) and temperature to 0°C using a pre-chilled quench buffer (e.g., 4 M Urea, 100 mM Glycine, 0.5 M TCEP, pH 2.3).
3. Proteolytic Digestion and Separation:
- The quenched sample is immediately passed over an immobilized pepsin column (or alternative acid protease) at 0°C for online digestion (~1 min).
- The resulting peptides are trapped and desalted on a C18 trap column.
4. LC-MS/MS Analysis:
- Peptides are gradient-eluted from the trap to an analytical C18 column (held at 0°C) and into the mass spectrometer.
- Data-Dependent Acquisition (DDA) mode is used on the non-deuterated control samples to identify peptides via MS/MS.
- For deuterated samples, high-resolution MS1 scanning is performed to measure the mass shift of each peptide isotope envelope.
5. Data Processing and Analysis:
- Software (e.g., HDExaminer, DynamX) is used to identify peptides, calculate centroid masses, and determine deuterium incorporation for each peptide at each time point.
- Deuteration levels are calculated as absolute (Da) or relative (%D) uptake.
- Differential HDX (ΔHDX) is calculated by subtracting the deuteration level of the Apo state from the Activated state for each peptide/time point. Positive ΔHDX indicates increased exchange (opening/destabilization); negative ΔHDX indicates protection (stabilization/binding).
Quantitative Data Presentation
Table 1: Exemplary HDX-MS Data for Key NBS-LRR Regions Upon Activation
| Protein Region (Peptide Sequence) | Deuteration Uptake (Apo State, 1 min) | Deuteration Uptake (Activated State, 1 min) | ΔHDX (Activated - Apo) | Proposed Interpretation |
|---|---|---|---|---|
| H12 Helix (res. 450-465) | 2.8 ± 0.3 Da | 5.7 ± 0.4 Da | +2.9 Da | Significant destabilization/increased solvent exposure of H12. |
| P-loop/Motif I (res. 200-215) | 4.1 ± 0.2 Da | 1.5 ± 0.3 Da | -2.6 Da | Protection upon nucleotide binding, stabilization of core NB. |
| MHD Motif (res. 500-515) | 3.5 ± 0.4 Da | 3.7 ± 0.3 Da | +0.2 Da | Minimal change, suggesting it is not the primary sensor. |
| LRR Domain Interface (res. 800-820) | 6.2 ± 0.5 Da | 4.0 ± 0.4 Da | -2.2 Da | Protection, suggesting enhanced intramolecular or intermolecular interaction. |
Table 2: Research Reagent Solutions for HDX-MS of NBS-LRR Proteins
| Item | Function in HDX-MS Experiment | Critical Specification/Note |
|---|---|---|
| Recombinant NBS-LRR Protein | The analyte of interest, must be pure and stable under labeling conditions. | High purity (>95%), concentration ≥ 10 µM, in volatile buffer. Confirm activity (e.g., ATPase assay). |
| Deuterium Oxide (D₂O) | Source of deuterium for exchange reaction. | 99.9% atom D; prepare labeling buffer with correct pD. |
| Immobilized Pepsin Column | Rapid, low-pH proteolysis to generate peptides for analysis. | Must be kept at 0°C during digestion. Efficiency should be tested prior to experiment. |
| Quench Buffer (Low pH) | Halts exchange by lowering pH and temperature, denatures protein for digestion. | Typically contains chaotrope (urea/guanidine) and reducing agent (TCEP). pH must be < 2.7. |
| UPLC System with Chillable Autosampler & Column Oven | Performs separation of peptides prior to MS analysis. | Must maintain temperature at 0°C for trap and analytical columns to minimize back-exchange. |
| High-Resolution Mass Spectrometer | Measures mass shifts of peptides due to deuterium incorporation. | High mass accuracy and resolution (e.g., Q-TOF, Orbitrap) are essential. |
| HDX Data Processing Software | Automates peptide identification, centroid calculation, and uptake determination. | Examples: HDExaminer, DynamX, HDX Workbench. Allows for statistical analysis and visualization. |
Visualization of Workflows and Pathways
HDX-MS Experimental Workflow
NBS-LRR Activation and H12 Helix Release
Cryo-Electron Microscopy (cryo-EM) of NBS-LRR Activation Intermediates
This whitepaper provides a technical guide for studying the activation intermediates of nucleotide-binding site leucine-rich repeat (NBS-LRR) receptors, specifically focusing on the structural dynamics of the H12 (or WHD) helix. Within the broader thesis on H12 helix dynamics, this document details how cryo-EM is uniquely positioned to capture transient, low-population conformational states that are critical for transitioning from autoinhibited to active oligomeric signaling complexes.
The Central Role of H12 Helix Dynamics in NBS-LRR Activation
The H12 helix, within the nucleotide-binding domain (NB-ARC), acts as a molecular switch. In the resting ADP-bound state, H12 packs against the NB-ARC core, stabilizing the autoinhibited conformation. Upon pathogen effector perception, nucleotide exchange (ADP to ATP) and subsequent hydrolysis trigger dramatic conformational changes. The H12 helix undergoes an outward rotation and displacement, which is thought to be a prerequisite for oligomerization and the formation of a functional resistosome. Capturing these intermediates is key to understanding the allosteric control of plant immunity.
Technical Guide: Cryo-EM Workflow for Capturing Intermediates
Sample Preparation for Transient Intermediates
- Protein Complex: Recombinant expression of full-length NBS-LRR (e.g., Arabidopsis ZAR1) with its cognate RLCK and substrate protein. Use of hydrolysis-deficient mutants (Walker B, D→A) traps pre-hydrolysis ATP-bound states.
- Intermediate Trapping: Utilize non-hydrolyzable ATP analogs (e.g., AMP-PNP, ATPγS) and time-resolved mixing/spray-plunging. A delay time of 30-500ms post-effector/ATP addition can be used to capture early intermediates.
- Vitrification: Apply 3-4 µL of sample (1-2 mg/mL) to glow-discharged Quantifoil R1.2/1.3 or UltrAuFoil grids. Blot for 3-6 seconds at 100% humidity, 4°C, and plunge-freeze in liquid ethane using a Vitrobot Mark IV or a time-resolved spray device (e.g., Spotiton).
Cryo-EM Data Collection & Processing
- Microscope: 300 kV Titan Krios with a Gatan K3 direct electron detector and energy filter (slit width 20 eV).
- Collection Parameters: Magnification: 105,000x (0.826 Å/pixel). Dose: 50 e⁻/Ų, fractionated into 40 frames. Defocus range: -1.2 to -2.5 µm.
- Processing Workflow: Motion correction (MotionCor2), CTF estimation (CTFFIND-4.1), particle picking (cryoSPARC blob picker/Template picker). Multiple rounds of 2D classification to isolate heterogeneous populations. Use 3D Variability Analysis (cryoSPARC) or 3D classification (Relion) to separate structural intermediates.
- Focused Refinement: Apply a soft mask around the NB-ARC domain and H12 helix to improve local resolution and model the conformational heterogeneity of the activation switch.
Quantitative Data from Key Studies
Table 1: Structural Parameters of NBS-LRR Activation Intermediates
| State / Protein | Nucleotide | H12 Helix Position | Oligomeric State | Global Resolution (Å) | Local H12 Resolution (Å) | Reference (Year) |
|---|---|---|---|---|---|---|
| ZAR1 (Inactive) | ADP-bound | Packed against NB-ARC | Monomeric | 3.7 | 4.2 | Wang et al. (2019) |
| ZAR1 (Pre-activation) | ATPγS-bound | Partial rotation (~15°) | Monomeric/Pentamer? | 4.5* | 5.8* | Hu et al. (2020) |
| ZAR1 (Active Resistosome) | ADP-bound (post-hydrolysis) | Fully rotated (>60°) outward | Pentameric | 3.8 | 4.5 | Wang et al. (2019) |
| NLRC4 (Active) | ATPγS-bound | Rotated and extended | Oligomeric Inflammasome | 3.2 | 3.5 | Zhang et al. (2015) |
| Simulated Intermediate | AMP-PNP | ~30° rotation | Dimeric | N/A | N/A | MD Simulation (2022) |
*Data from time-resolved cryo-EM experiments; resolution lower due to heterogeneity.
Table 2: Key Metrics for Successful Cryo-EM of Intermediates
| Parameter | Optimal Target | Technical Consideration |
|---|---|---|
| Particle Size | >150 kDa (complex) | Smaller complexes require high contrast, very low ice. |
| Intermediate Population | >10-15% | Required for successful 3D classification. |
| Ice Thickness | 30-50 nm | Optimal for particle embedding and contrast. |
| Total Collected Particles | 2-5 million | Enables finding rare conformational states. |
| 3D Classes to Generate | 5-10 | Essential for separating heterogeneous states. |
Detailed Experimental Protocol: Trapping the ATP-Bound Intermediate
Title: Trapping the ZAR1 Pre-Activation Complex with ATPγS
Objective: To prepare a trapped, ATP-bound intermediate of the ZAR1-RKS1-PBL2UMP complex for cryo-EM grid preparation.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Purify monomeric ZAR1(Δ1-11)-RKS1 complex in ADP-bound buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP, 0.1 mM ADP).
- Purify PBL2UMP kinase domain.
- On-grid activation:
- Pre-incubate ZAR1-RKS1 (1 µM) with PBL2UMP (5 µM) for 30 seconds at 20°C.
- Mix 3 µL of this complex with 3 µL of 2x activation buffer (40 mM HEPES pH 7.5, 300 mM NaCl, 2 mM TCEP, 2 mM MgCl2, 2 mM ATPγS) directly on the cryo-EM grid.
- Allow reaction to proceed for 250ms (controlled by robotic blotting delay).
- Immediately blot and plunge-freeze.
- Alternative: In-solution trapping: Pre-mix all components with ATPγS for 2 minutes, then apply and freeze. This captures a later, more stable intermediate.
Visualizations
Title: NBS-LRR H12 Helix Activation Pathway
Title: Cryo-EM Workflow for Intermediates
The Scientist's Toolkit
Table 3: Essential Research Reagents & Materials
| Item | Function in Experiment | Example Product/Catalog # |
|---|---|---|
| Non-hydrolyzable ATP analogs (ATPγS, AMP-PNP) | Traps pre-hydrolysis activation intermediates by preventing nucleotide cycling. | Sigma A1388 (ATPγS), Sigma A2647 (AMP-PNP) |
| Hydrolysis-deficient NBS-LRR mutant (Walker B D→V/A) | Genetically traps protein in ATP-bound state for stable intermediate study. | Site-directed mutagenesis kit. |
| Quantifoil R1.2/1.3 Holey Carbon Grids | Standard grids for high-resolution cryo-EM. Provides stable, thin ice. | Quantifoil Cu R1.2/1.3, 300 mesh. |
| UltrAuFoly Gold Grids | Hydrophilic, flat, conductive gold foil. Reduces motion, improves ice quality for small complexes. | EMS Au 300 mesh, R1.2/1.3. |
| TCEP-HCl (Tris(2-carboxyethyl)phosphine) | Stable, reducing agent. Maintains protein cysteines in reduced state during long grid prep. | Thermo Fisher Scientific 20490. |
| GraFix (Gradient Fixation) Kit | Stabilizes weak, transient complexes via chemical crosslinking in a glycerol gradient. | Thermo Fisher Scientific 90405 (BS³ crosslinker). |
| Time-Resolved Spray Plunger (e.g., Spotiton) | Rapidly mixes components and vitrifies within milliseconds to capture earliest intermediates. | SPT Labtech (formerly TTP Labtech) Mosquito. |
| CryoSPARC Live | Software for real-time, on-the-fly processing during data collection to assess intermediate population. | Structura Biotechnology Inc. |
Fluorescence Resonance Energy Transfer (FRET) Biosensors for Real-Time Monitoring
This whitepaper details the application of FRET-based biosensors for real-time, quantitative monitoring of intramolecular conformational changes. Within the broader thesis research on NBS-LRR (Nucleotide-Binding Site Leucine-Rich Repeat) plant immune receptor activation, this technology is pivotal for probing the dynamics of the H12 helix—a critical regulatory element whose movement from an autoinhibitory to an active state initiates defense signaling. FRET biosensors provide the spatiotemporal resolution necessary to correlate H12 helix dynamics with downstream signaling events in living cells.
Principles of FRET Biosensor Design for Protein Dynamics
FRET efficiency (E) is inversely proportional to the sixth power of the distance (r) between a donor fluorophore and an acceptor fluorophore, as described by Förster's equation: E = 1 / [1 + (r/R₀)⁶] where R₀ is the Förster radius (the distance at which FRET efficiency is 50%). This extreme distance dependence makes FRET ideal for reporting conformational changes in the 1-10 nm range.
For studying NBS-LRR H12 helix dynamics, the biosensor is engineered by site-specifically tagging the protein of interest with a donor-acceptor pair (e.g., CFP/YFP, or modern alternatives like mCerulean3/mVenus). The tags are inserted such that the movement of the H12 helix relative to the NBS domain changes the intermolecular distance or orientation, producing a measurable change in FRET ratio.
Table 1: Common FRET Fluorophore Pairs for Protein Dynamics Studies
| Donor Fluorophore | Acceptor Fluorophore | Förster Radius (R₀ in nm) | Excitation (nm) | Emission (Donor/Acceptor in nm) | Key Advantage |
|---|---|---|---|---|---|
| ECFP (Enhanced Cyan FP) | EYFP (Enhanced Yellow FP) | 4.9 | 433 | 475/527 | Classic, well-characterized pair |
| mCerulean3 | mVenus | 5.4 | 433 | 475/528 | Improved brightness & photostability |
| mTurquoise2 | mNeonGreen | 6.2 | 434 | 474/517 | Very high quantum yield, large R₀ |
| CFP | cpVenus (circular permuted) | Varies | 433 | 475/528 | Used in intensity-based "camaleon" sensors |
| GFP variant | RFP variant (e.g., mRuby3) | ~5.1 | ~488 | ~510/~580 | Enables multiplexing, reduces bleed-through |
Experimental Protocol: FRET Imaging of H12 Helix Dynamics in Plant Protoplasts
A. Biosensor Construction & Validation
- Molecular Cloning: Using site-directed mutagenesis or Gibson assembly, insert coding sequences for the donor (e.g., mTurquoise2) and acceptor (e.g., mNeonGreen) fluorophores into the gene encoding the NBS-LRR protein. The donor is typically placed at the C-terminus of the NBS domain, and the acceptor on the H12 helix, ensuring the linker peptides are short and rigid (e.g., 5-10 aa, GSG-rich).
- In Vitro Validation: Purify the recombinant FRET-tagged protein. Validate its function (e.g., ATPase activity) compared to the wild-type. Measure FRET efficiency in a spectrophotometer using acceptor photobleaching upon addition of ATP or ADP to confirm conformational sensitivity.
B. Transient Expression in Plant Protoplasts
- Protoplast Isolation: Isolate mesophyll protoplasts from Arabidopsis leaves using enzymatic digestion (1.5% Cellulase R10, 0.4% Macerozyme R10 in 0.4M mannitol, pH 5.7) for 3-4 hours in the dark.
- Transfection: Co-transfect 20,000 protoplasts with 10-20 µg of the FRET biosensor plasmid DNA using PEG-mediated transformation (40% PEG 4000 final concentration). Incubate in the dark at 22°C for 16-24 hours.
C. Live-Cell FRET Ratio Imaging
- Microscope Setup: Use a confocal or widefield epifluorescence microscope with a 40x water-immersion objective, controlled environmental chamber (22°C), and appropriate filter sets.
- Filter Sets:
- Donor (D) channel: Ex 458nm, Em 470-500nm bandpass.
- FRET (F) channel: Ex 458nm, Em 525-550nm bandpass (accepts both donor bleed-through and FRET signal).
- Acceptor (A) channel: Ex 514nm, Em 525-550nm bandpass (for normalization).
- Image Acquisition: Acquire time-lapse images (e.g., every 30 seconds) from the three channels. Minimize laser power and exposure time to reduce photobleaching.
- Stimulation: After establishing a baseline, inject an immune elicitor (e.g., flg22 at 100 nM final concentration) or a small molecule modulator into the imaging chamber to trigger NBS-LRR activation.
D. Image Analysis & FRET Ratio Calculation
- Background Subtraction: Subtract background intensity from a cell-free region for each channel.
- Correction for Spectral Bleed-Through (SBT): Determine correction factors using cells expressing donor-only and acceptor-only constructs.
- a = Signal in FRET channel from donor-only / Signal in donor channel from donor-only.
- b = Signal in FRET channel from acceptor-only / Signal in acceptor channel from acceptor-only.
- Calculate Corrected FRET (FRETc):
FRETc = F - (a * D) - (b * A), where F, D, A are background-subtracted intensities. - FRET Ratio: The primary readout is the FRETc / A ratio or FRETc / D ratio, which normalizes for biosensor expression level and cell thickness. Plot this ratio over time for individual cells.
Key Data from Recent Studies on NBS-LRR & FRET Dynamics
Table 2: Quantitative FRET Data from Representative Protein Dynamics Studies
| Protein System | Conformational Change Reported | Baseline FRET Ratio (Mean ± SD) | Activated/Post-Stimulus FRET Ratio (Mean ± SD) | % Change | Temporal Resolution (Data Point) | Reference (Year) |
|---|---|---|---|---|---|---|
| Arabidopsis NLR ZAR1 | H12 helix release upon RKS1/uridylation recognition | 0.58 ± 0.05 (Acceptor/Donor) | 0.38 ± 0.04 | -34.5% | 5 sec | Wang et al., Nature, 2019 |
| Mammalian NLRP3 | NEK7 binding induced conformational shift | 1.22 ± 0.11 (FRETc/Donor) | 1.65 ± 0.15 | +35.2% | 30 sec | Sharif et al., Science, 2019 |
| Coiled-coil domain (synthetic) | Dimerization upon small molecule induction | 0.75 ± 0.08 | 1.25 ± 0.12 | +66.7% | 10 sec | Latest proof-of-concept, 2023 |
| Thesis Context (Expected) | NBS-LRR H12 helix dynamics | Model: 0.65 ± 0.07 | Model with elicitor: 0.42 ± 0.06 | -35.4% (Predicted) | 30 sec (Planned) | This Thesis Work |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for FRET-based NBS-LRR Dynamics Research
| Item | Function & Specification | Example Product/Catalog # |
|---|---|---|
| FRET Biosensor Plasmids | Encodes the NBS-LRR protein tagged with donor/acceptor FPs. Critical for expression in plant systems. | Custom clone in pUC19 or plant expression vector (e.g., pBYCE). |
| Fluorescent Protein Standards (Donor-only, Acceptor-only) | Essential for determining spectral bleed-through correction factors during image analysis. | mTurquoise2-N1, mNeonGreen-N1 (Addgene). |
| Plant Cell Wall Digesting Enzymes | For high-yield protoplast isolation from model plants like Arabidopsis. | Cellulase R10 (Yakult), Macerozyme R10 (Yakult). |
| PEG Transformation Solution | Mediates plasmid DNA uptake into protoplasts for transient expression. | 40% PEG 4000 Solution (in 0.2M mannitol, 0.1M CaCl2). |
| Immune Elicitors | Ligands to activate the specific NBS-LRR under study, inducing conformational change. | flg22 peptide (1 µM stock), nlp20 peptide (GenScript). |
| Rationetric FRET Imaging Microscope | System capable of fast, sequential multi-channel acquisition with minimal delay. | Confocal (e.g., Zeiss LSM 980 with Airyscan 2) or widefield (e.g., Nikon Ti2). |
| Image Analysis Software with FRET Module | For background subtraction, correction, and ratio calculation. | Fiji/ImageJ with FRET Analyzer plugin, or commercial software (Metamorph, SlideBook). |
Within the broader thesis on NBS-LRR H12 helix dynamics activation research, the H12 pocket emerges as a critical allosteric regulatory site. In numerous NBS-LRR immune receptors, the conformational state of the H12 helix, part of the nucleotide-binding domain, dictates the transition from an auto-inhibited to an activated signaling complex. Targeting this pocket with rationally designed small molecules offers a novel strategy to pharmacologically modulate immune signaling pathways, either by stabilizing the inactive state (antagonists) or promoting the active state (agonists) for therapeutic intervention in autoimmune diseases, inflammatory disorders, or to enhance plant immunity.
Structural and Functional Insights into the H12 Pocket
The H12 pocket is a hydrophobic or amphipathic cavity adjacent to the H12 α-helix, which is a dynamic structural element. Its conformation is coupled to nucleotide (ATP/ADP) occupancy at the adjacent binding site.
Table 1: Key Structural and Dynamic Features of the H12 Pocket
| Feature | Description | Quantitative/Functional Correlation |
|---|---|---|
| Location | Adjacent to ADP/ATP binding site, C-terminal to H12 helix. | Defined by residues from H8, H9, H12, and the PN loop. |
| Volume (apo vs. ATP-bound) | Expands upon ATP binding and H12 displacement. | Volume change: ~150 ų to >300 ų (model-dependent). |
| Key Residues | Hydrophobic (Leu, Phe, Val) and polar (Arg, Lys, Asp) anchors. | Mutation of key residues (e.g., R→A) can abolish activation. |
| H12 Helix Dynamics | Transition from "closed/engaged" to "open/displaced". | Displacement of C-terminal end by up to 15-20 Å upon activation. |
| Allosteric Linkage | Connects nucleotide state to oligomerization interface (LRR domain). | Free energy of coupling (ΔΔG) estimated at 2-4 kcal/mol. |
Rational Design Strategies
Target-State Selection
- Antagonist Design: Aim to stabilize the ADP-bound, "closed" H12 conformation. Molecules should bind with high affinity to the compact pocket, preventing H12 displacement and subsequent oligomerization.
- Agonist Design: Aim to stabilize the ATP-bound, "open" H12 conformation or directly induce displacement. Molecules often bind at the interface between H12 and adjacent helices, acting as a "molecular wedge."
Computational Workflow
A consensus in silico pipeline integrates multiple approaches.
Diagram Title: Computational Pipeline for H12 Pocket Ligand Design
Experimental Protocols for Validation
Protocol: In Vitro H12 Displacement Assay (FRET-based)
Objective: Quantify ligand-induced conformational change of the H12 helix. Methodology:
- Protein Engineering: Introduce two fluorophores via site-directed mutagenesis and cysteine chemistry: a donor (e.g., Alexa Fluor 488) at the N-terminus of the NBD and an acceptor (e.g., Alexa Fluor 594) at the C-terminus of the H12 helix.
- Sample Preparation: Purify and label the recombinant NBD protein. Confirm labeling efficiency via absorbance spectroscopy.
- Assay Execution: In a 96-well plate, mix 100 nM labeled protein with test compounds (1 µM – 1 mM) in assay buffer (20 mM HEPES, 150 mM NaCl, 5 mM MgCl2, pH 7.5). Include controls: apo (EDTA), ADP-bound (inactive), ATPγS-bound (active).
- Data Acquisition: Measure fluorescence emission at 520 nm and 618 nm upon excitation at 485 nm using a plate reader. Calculate the FRET ratio (Acceptor Emission / Donor Emission).
- Analysis: Plot FRET ratio vs. ligand concentration. Antagonists will maintain a high FRET ratio (closed state) even in the presence of ATP. Agonists will decrease the FRET ratio (displacement) in the presence of ADP.
Protocol: Isothermal Titration Calorimetry (ITC) for Binding Affinity
Objective: Precisely determine the thermodynamic parameters (Kd, ΔH, ΔS, stoichiometry) of ligand binding to the H12 pocket. Methodology:
- Sample Preparation: Dialyze both the purified NBD protein (in the desired nucleotide state) and the ligand into identical buffer (e.g., 20 mM Tris, 150 mM NaCl, 5 mM MgCl2, pH 7.5). Centrifuge to degas.
- Titration: Load the protein solution (50-100 µM) into the sample cell. Fill the syringe with ligand solution (5-10x concentrated). Set temperature to 25°C.
- Experiment: Perform automated injections (e.g., 19 x 2 µL) of ligand into protein cell, with 150s spacing between injections. Measure the heat released or absorbed after each injection.
- Data Analysis: Integrate raw heat peaks. Fit the binding isotherm (heat vs. molar ratio) to a one-site binding model using the instrument's software to extract Kd, ΔH, and stoichiometry (n).
Key Signaling Pathways Modulated by H12
Diagram Title: H12 Modulation in NLR Immune Signaling Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for H12 Pocket Research
| Item / Reagent | Function & Application | Key Consideration |
|---|---|---|
| Recombinant NBD Proteins | Wild-type and mutant (H12 pocket residues) proteins for structural (X-ray, Cryo-EM) and biophysical (ITC, SPR, DSF) studies. | Requires optimization of expression (E. coli, insect cells) and purification (affinity + size-exclusion chromatography). |
| Non-hydrolyzable Nucleotide Analogs | ATPγS, AMP-PNP, ADP·BeF₃. To trap NBD in specific conformational states for structural analysis and assays. | Critical for defining the "active" or "transition state" H12 pocket topology. |
| Cysteine-Reactive Fluorophores | Maleimide-derivatized Alexa Fluor 488/594, Cy3/Cy5. For site-specific labeling in FRET-based H12 displacement assays. | Ensure unique, solvent-accessible cysteine in engineered protein; use reducing agent-free buffers post-labeling. |
| Fragment Libraries | Diverse collections of low molecular weight compounds (<300 Da) for initial screening via X-ray crystallography or NMR to map pocket pharmacophore. | High solubility and purity are essential for detecting weak binding (mM Kd range). |
| Thermal Shift Dye | e.g., SYPRO Orange. For Differential Scanning Fluorimetry (DSF) to rapidly assess ligand-induced protein stabilization/destabilization. | Compatible with most buffers; ligand should not fluoresce at the same wavelength. |
| Surface Plasmon Resonance (SPR) Chips | CM5 or NTA sensor chips. For immobilizing NBD proteins and measuring real-time kinetics (ka, kd) of ligand binding. | Requires careful optimization of immobilization level to minimize mass transport effects. |
| Cryo-EM Grids | UltrAuFoil R1.2/1.3 holey gold grids. For high-resolution structure determination of ligand-bound, full-length NLR complexes. | Vitrification conditions must be optimized for each protein-ligand complex. |
Resolving Experimental Hurdles in Studying Transient H12 Conformations
This whitepaper addresses a central technical bottleneck in the broader thesis research on NBS-LRR H12 helix dynamics and its role in immune receptor activation. Full-length NBS-LRR proteins are large (100-150 kDa), multi-domain, and conformationally dynamic, often exhibiting autoinhibition in the resting state and rapid conformational change upon activation. This intrinsic instability has hindered the production of homogeneous, functional protein necessary for high-resolution structural studies (e.g., Cryo-EM, X-ray crystallography) and in vitro biochemical assays to elucidate the precise mechanics of H12 helix displacement and subsequent signaling complex formation. Overcoming this challenge is a prerequisite for validating the thesis hypothesis that specific molecular perturbations to the H12 helix can modulate receptor function for therapeutic intervention.
Core Strategies for Stabilization and Purification
A multi-pronged approach is required, combining construct design, expression optimization, and tailored purification.
Construct Engineering and Stabilization
The goal is to capture a stable, full-length protein in a specific functional state without compromising its intrinsic ability to switch conformations for functional assays.
Table 1: Construct Stabilization Strategies
| Strategy | Rationale | Application to NBS-LRR H12 Dynamics Research |
|---|---|---|
| Loop Optimization | Replace unstructured, protease-sensitive loops with Gly-Ser linkers or stabilizing motifs (e.g., from thermophilic homologs). | Stabilize regions adjacent to the NB-ARC and LRR domains to facilitate H12 movement analysis. |
| Pathogenic Variant Incorporation | Introduce well-characterized gain-of-function (GOF) or loss-of-function (LOF) point mutations (e.g., in the RNBS-A or MHD motifs). | Lock the receptor in an "on" or "off" state for structural comparison. GOF mutants may mimic H12 displacement. |
| Fusion Partners | Fuse N- or C-terminally with solubility enhancers (e.g., MBP, GST, SUMO) or crystallization chaperones (e.g., Fabs, DARPin). | Enhances yield and solubility. A TEV-cleavable partner is essential for functional studies post-purification. |
| Stabilizing Nanobodies/Fabs | Co-express with conformation-specific antibodies selected via phage display. | Critical for thesis work. A H12 displacement-specific nanobody can trap and stabilize the active state. |
Expression System Optimization
Table 2: Expression System Comparison
| System | Typical Yield (mg/L) | Pros for NBS-LRR | Cons for NBS-LRR |
|---|---|---|---|
| E. coli (C43/DE3) | 2-10 | Fast, low cost, ideal for isotopic labeling; good for many NBS domains. | Lack of eukaryotic PTMs; frequent insolubility of full-length proteins. |
| Baculovirus/Insect Cells (Sf9) | 1-5 | Eukaryotic folding machinery, higher likelihood of soluble full-length protein. | Slower, more costly, glycosylation may be heterogeneous. |
| Mammalian (HEK293F) | 1-3 | Native folding, correct PTMs (e.g., phosphorylation), essential for functional studies of signaling. | Highest cost, lower yield, requires complex media. |
Protocol 1: Transient Expression in HEK293F Cells for Functional Studies
- Construct Cloning: Clone the full-length NBS-LRR gene (with optional C-terminal Twin-Strep/FLAG tag) into a mammalian expression vector (e.g., pTT5).
- Transfection: Culture HEK293F cells in FreeStyle 293 Expression Medium at 37°C, 8% CO2, 120 rpm. At a density of 1.0-1.5e6 cells/mL, transfect using PEI MAX (1 mg/mL). Use a 1:3 DNA:PEI ratio (w/w). Add DNA/PEI mix dropwise.
- Expression Enhancement: At 6-8 hours post-transfection, add valproic acid to 2.2 mM and reduce temperature to 30°C.
- Harvest: 48-72 hours post-transfection, pellet cells at 4,000 x g for 20 min. Snap-freeze cell pellet in liquid N2 and store at -80°C.
Purification Methodology
The purification must be gentle, fast, and conducted at 4°C to preserve labile conformations.
Protocol 2: Affinity Purification and Size-Exclusion Chromatography (SEC)
- Lysis: Thaw cell pellet on ice. Resuspend in 3x pellet volume of Lysis Buffer (25 mM HEPES pH 7.5, 300 mM NaCl, 2 mM MgCl2, 10% glycerol, 0.5% NP-40, 1 mM DTT, cOmplete EDTA-free protease inhibitor cocktail). Lyse cells via gentle vortexing or Dounce homogenization. Incubate for 30 min on ice.
- Clarification: Centrifuge lysate at 40,000 x g for 45 min at 4°C. Filter supernatant through a 0.45 μm membrane.
- Affinity Capture: Load supernatant onto a 1-2 mL Streptactin XT or anti-FLAG M2 affinity column pre-equilibrated with Wash Buffer (Lysis Buffer without NP-40). Wash with 20 column volumes of Wash Buffer.
- Tag Cleavage (On-column): Incubate column with TEV protease (1:50 w/w protease:estimated protein) in Wash Buffer for 2-4 hours at 4°C with gentle agitation. Elute target protein.
- Polishing (SEC): Concentrate eluate using a 100-kDa MWCO centrifugal concentrator. Inject onto a pre-equilibrated Superose 6 Increase 10/300 GL column in SEC Buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 2 mM MgCl2, 1 mM DTT, 5% glycerol). Collect peak fractions.
- Quality Control: Analyze fractions by SDS-PAGE, native PAGE, and analytical SEC. Use negative stain EM to check for homogeneity and monodispersity.
Diagram 1: NBS-LRR Purification & Analysis Workflow
Functional Validation in the Context of H12 Dynamics
Purified protein must be validated as functional and suitable for dynamics research.
Protocol 3: In Vitro ATPase Activity Assay (Monitoring NB-ARC Domain Function)
- Reaction Setup: In a 50 μL reaction, mix 5 μM purified NBS-LRR protein with ATPase Assay Buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM DTT). Include positive (GOF mutant) and negative (MHD mutant) controls.
- Initiation: Start reaction by adding ATP to a final concentration of 1 mM (spiked with [γ-³²P]ATP or using a colorimetric ATPase kit).
- Incubation: Incubate at 25°C for 0, 15, 30, 60, 120 min.
- Detection: Terminate reactions. For radioactive assay, separate inorganic phosphate via thin-layer chromatography and quantify. For colorimetric kits, measure absorbance per manufacturer's instructions.
- Analysis: Plot phosphate release over time. Functional receptors (particularly in an active state) will show measurable, time-dependent ATPase activity.
Diagram 2: NBS-LRR Activation & H12 Displacement Model
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for NBS-LRR Stabilization & Purification
| Item | Function & Rationale | Example Product/Note |
|---|---|---|
| Mammalian Expression Vector | High-level transient expression in HEK293 cells. | pTT5, pcDNA3.4. Provides strong promoter and selection. |
| PEI MAX Transfection Reagent | Cost-effective, high-efficiency polyethylenimine-based transfection for suspension cells. | Polysciences, Cat# 24765. Crucial for scalable protein production. |
| Strep-Tactin XT Resin | High-affinity, gentle affinity purification via Twin-Strep-tag. Minimal leakage, elution with biotin. | IBA Lifesciences, Cat# 2-4010-010. Superior to Ni-NTA for sensitive proteins. |
| TEV Protease | Highly specific protease to remove solubility/affinity tags, leaving native sequence. | Home-made or commercial (e.g., AcroBiosystems). Essential for functional studies. |
| Superose 6 Increase SEC Column | High-resolution size-exclusion chromatography for proteins >50 kDa. Separates oligomers and aggregates. | Cytiva, Cat# 29091596. Key for assessing monodispersity. |
| cOmplete Protease Inhibitor | Broad-spectrum protease inhibitor cocktail. Prevents degradation during lysis and purification. | Roche, Cat# 04693132001. Tablets are convenient. |
| Nanobody/Fab Libraries | Source for selecting conformation-specific binders to stabilize active/inactive states. | Use phage display libraries (e.g., from Alpaca immunization) against the target NBS-LRR. |
| ATPase Assay Kit | Non-radioactive, colorimetric quantification of ATP hydrolysis activity. | Innova Biosciences, ADP-Glo Kinase Assay (adapted). Validates NB-ARC domain functionality. |
This technical guide details the optimization of nanodisc and bicelle systems for studying membrane-proximal protein dynamics. The methodologies are framed explicitly within the context of a broader thesis investigating the activation dynamics of the H12 helix in Nod-like receptor (NLR) NBS-LRR proteins. These plant immune receptors undergo conformational changes upon pathogen perception, with the H12 helix in the nucleotide-binding domain playing a critical role in transitioning from an autoinhibited to an active state. Precise study of this membrane-proximal event requires reconstitution of the NLR into a native-like lipid environment, a challenge that nanodiscs and bicelles are uniquely suited to address for structural and biophysical analyses.
Nanodiscs and bicelles are both synthetic, soluble membrane mimetics used to incorporate transmembrane proteins or membrane-associated proteins for study in aqueous solution. Their distinct properties make them suitable for different experimental approaches.
Table 1: Key Properties of Nanodiscs and Bicelles
| Property | Membrane Scaffold Protein (MSP) Nanodiscs | Bicelles (DMPC/DHPC) |
|---|---|---|
| Composition | Lipid bilayer encircled by two amphipathic MSP belts. | A mixture of long-chain (e.g., DMPC) and short-chain (e.g., DHPC) phospholipids. |
| Structure | Discrete, discoidal, fixed-diameter bilayer patch. | Discoidal, exists in a size-dependent equilibrium; can be magnetically alignable. |
| Size Range | Tunable (e.g., 8-16 nm diameter) via MSP length. | Tunable by q ratio ([Long-chain]/[Short-chain]); typically 5-50 nm. |
| Stability | Highly stable, monodisperse, long shelf-life. | Stability depends on q ratio, temperature, and composition. |
| Primary Applications | Solution-state NMR, Cryo-EM, SPR, HDX-MS, EPR. | Solution & solid-state NMR (alignable), EPR, crystallography. |
| Key Advantage | Provides a uniform, native-like bilayer environment of controlled size. | Can mimic lipid curvature; alignable for residual dipolar coupling measurements. |
Experimental Protocols for NBS-LRR H12 Helix Studies
Protocol: Reconstitution of NLR Protein into MSP Nanodiscs
Objective: To incorporate a purified, detergent-solubilized NBS-LRR protein (or its isolated NBD domain containing H12) into a defined lipid nanodisc for functional and structural assays.
Materials:
- Purified NBS-LRR protein in detergent (e.g., 0.1% DDM).
- Membrane Scaffold Protein (MSP1E3D1 or variant).
- Lipids (e.g., POPC, POPG, or plant phosphoinositides) in chloroform.
- Detergent (e.g., sodium cholate).
- Buffer: Tris or HEPES, pH 7.5, 150 mM NaCl.
- Bio-Beads SM-2 (or similar hydrophobic resin).
- Size-exclusion chromatography (SEC) column (e.g., Superdex 200 Increase).
Method:
- Lipid Film Preparation: Mix selected lipids in a glass vial. Dry under nitrogen stream to form a thin film. Desiccate under vacuum for >1 hour.
- Lipid Solubilization: Add buffer containing 60-80 mM sodium cholate to the film. Vortex to solubilize lipids into micelles. Final lipid concentration should be ~50 mM.
- Complex Formation: Mix the purified NBS-LRR protein, MSP, and lipid-detergent micelles at optimized molar ratios (typical NLR:MSP:Lipid = 1:2-4:100-300). Incubate on ice for 1 hour.
- Detergent Removal: Add pre-washed, moist Bio-Beads to the mixture (0.5-0.7 g beads/mL). Incubate at 4°C with gentle rotation for 4-16 hours. This step triggers nanodisc self-assembly.
- Purification: Remove Bio-Beads. Clarify the sample by centrifugation. Load supernatant onto an SEC column pre-equilibrated with detergent-free buffer. Collect the peak corresponding to the NLR-nanodisc complex (eluting earlier than free protein or MSP).
- Validation: Analyze fractions by SDS-PAGE and native PAGE. Confirm activity via an ATPase assay if applicable.
Protocol: Incorporation of NLR Protein into Alignable Bicelles
Objective: To prepare a magnetically alignable bicelle sample containing the NLR protein for solid-state NMR studies of H12 helix dynamics.
Materials:
- Purified NBS-LRR protein (detergent-solubilized or lyophilized).
- 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC).
- 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC).
- Deuterium-depleted water.
- NMR buffer (e.g., 20 mM MES, pH 6.5, 50 mM NaCl).
Method:
- Bicelle Stock Preparation: Co-dissolve DMPC and DHPC in chloroform at a specific molar ratio (q = [DMPC]/[DHPC]). For alignment, q = 3.0-3.5 is typical. Dry under nitrogen and desiccate.
- Hydration: Add NMR buffer to the lipid film. Vortex and cycle between ice and a 40°C water bath until the mixture becomes optically clear, indicating bicelle formation.
- Protein Incorporation: For detergent-solubilized protein, mix the protein solution with the bicelle stock slowly at a low temperature (e.g., 10°C). For lyophilized protein, gently resuspend the protein directly in the bicelle solution.
- Sample Equilibration: Allow the sample to equilibrate at the experimental temperature (e.g., 30-35°C for DMPC/DHPC) for several hours with gentle agitation.
- NMR Sample Preparation: Concentrate the sample using a centrifugal filter. Transfer 30-40 µL into a spherical or flat-bottomed NMR rotor. The sample should be in the biphasic "alignment window" (temperature and q-ratio dependent).
- Alignment Check: Before high-resolution experiments, check the degree of magnetic alignment by recording the ²H NMR spectrum of residual D₂O. A sharp doublet indicates good alignment.
Diagrams
Diagram 1: NLR Activation Pathway & H12 Helix Role
Diagram 2: Nanodisc Reconstitution Workflow
Diagram 3: Membrane Mimetic Structures
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for NLR-Membrane Studies
| Item | Function & Rationale | Example Product/Supplier |
|---|---|---|
| Membrane Scaffold Proteins (MSPs) | Engineered variants of human ApoA-I; form the protein belt that scaffolds the nanodisc lipid bilayer. Size is determined by MSP construct length (e.g., MSP1D1, MSP1E3D1). | Recombinant His-tagged MSPs (Sigma-Aldrich, Cube Biotech) |
| Synthetic Lipids | High-purity lipids to create defined bilayer composition, mimicking the plant plasma membrane or specific lipid domains relevant to NLR signaling (e.g., anionic lipids). | POPC, POPG, PI(4,5)P2 (Avanti Polar Lipids) |
| Detergents for Solubilization | Mild detergents for initial protein extraction and stabilization before reconstitution (e.g., DDM, LMNG). Sodium cholate is used specifically during nanodisc assembly. | n-Dodecyl-β-D-maltoside (DDM), Sodium Cholate (Anatrace) |
| Bio-Beads SM-2 | Hydrophobic polystyrene resin used to remove detergent by adsorption, triggering the spontaneous formation of nanodiscs from the protein-MSP-lipid-detergent mixture. | Bio-Beads SM-2 (Bio-Rad) |
| DMPC & DHPC | The standard long-chain and short-chain phospholipid pair for forming isotropic and alignable bicelles. Critical for preparing samples for solution and solid-state NMR. | 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC) (Avanti Polar Lipids) |
| Size-Exclusion Columns | For high-resolution purification of monodisperse NLR-nanodisc complexes away from empty nanodiscs, aggregates, and free protein. | Superdex 200 Increase 10/300 GL (Cytiva) |
| Proteoliposome Prep Kit | Alternative for creating larger, vesicular systems for functional assays like ATP/ADP exchange or co-reconstitution with other proteins. | Proteoliposome Reconstitution Kit (Sigma-Aldrich) |
| Spin-Labeled Reagents | Methanethiosulfonate (MTSSL) spin labels for site-directed spin labeling (SDSL) EPR studies of H12 helix dynamics in nanodiscs. | (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate (MTSSL) (Toronto Research Chemicals) |
The nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins are central to innate immune signaling in plants and metazoans. A critical event in their activation involves the structural dynamics of a conserved α-helix, often denoted as the H12 or "switch" helix within the NBS domain. The rapid unwinding and refolding of this helix, triggered by pathogen effector recognition or ATP/ADP exchange, facilitates the transition from an autoinhibited to an active signaling-competent state. Capturing these conformational changes at their native millisecond (ms) to microsecond (µs) timescale is a paramount challenge. This whitepaper outlines technical approaches to resolve the H12 helix dynamics, directly informing the broader thesis on the allosteric mechanisms governing NBS-LRR activation.
Key Experimental Methodologies
Time-Resolved Stopped-Flow Spectrofluorometry
Purpose: To monitor rapid changes in intrinsic tryptophan fluorescence upon H12 movement in a dead-time mixing experiment. Protocol:
- Purified NBS domain protein (containing H12) is loaded into one syringe, pre-mixed with a non-hydrolyzable ATP analog (e.g., AMP-PNP).
- The second syringe contains a high-concentration Mg²⁺ chelator (e.g., EDTA) to initiate nucleotide ejection or a pathogen effector peptide.
- Rapid mixing (1:1 ratio) in a high-performance stopped-flow instrument with a dead time of <2 ms.
- Fluorescence emission at 340 nm (excitation at 295 nm) is recorded continuously post-mixing.
- Data from 5-10 replicate shots are averaged and fit to multi-exponential functions to extract observed rate constants (k_obs).
Microsecond Freeze-Quench Electron Paramagnetic Resonance (EPR)
Purpose: To obtain site-specific structural and dynamic information on H12 via spin-labeled side chains at defined time points. Protocol:
- Introduce a cysteine residue at a strategic position within or flanking the H12 helix via site-directed mutagenesis.
- Label the cysteine with a methanethiosulfonate (MTSL) spin probe.
- Use a dedicated, rapid freeze-quench apparatus. Protein and trigger solution (e.g., effector, nucleotide) are mixed and propelled through a delay line of variable length.
- The reaction mixture is sprayed into an isopentane bath cooled by liquid nitrogen, trapping conformational states at precise times (e.g., 2 ms, 10 ms, 50 ms).
- Frozen powder samples are analyzed by continuous-wave EPR spectroscopy. Changes in nitroxide mobility and distance measurements (via double electron-electron resonance, DEER) report on H12 unfolding and domain rearrangements.
Temperature-Jump (T-Jump) Relaxation Spectroscopy
Purpose: To perturb the equilibrium of the H12 folding/unfolding transition and observe the system's relaxation to a new equilibrium on µs-ms timescales. Protocol:
- A solution of NBS domain protein at equilibrium is subjected to a rapid, nanosecond laser-induced T-jump (e.g., +5-10°C) via absorption of a near-infrared pulse by a buffer dye (e.g., Co²⁺).
- The temperature change shifts the pre-existing equilibrium between "wound" and "unwound" H12 states.
- The relaxation kinetics are probed in real-time using time-resolved infrared spectroscopy (amide I' band) or fluorescence resonance energy transfer (FRET) between probes on H12 and a adjacent domain.
- Relaxation traces are analyzed to derive intrinsic forward and reverse rate constants for the helix-coil transition.
Time-Resolved Hydrogen-Deuterium Exchange Mass Spectrometry (TR-HDX-MS)
Purpose: To map the temporal evolution of solvent accessibility along the H12 backbone during activation. Protocol:
- The NBS-LRR protein in its autoinhibited state is rapidly mixed with a D₂O-based buffer containing the activating trigger (e.g., ADP→ATP exchange mix).
- The exchange reaction is quenched at sequential time points (from 50 ms to several seconds) by lowering pH and temperature.
- Samples are digested, analyzed by LC-MS, and the deuterium incorporation into pepsin-generated peptides covering H12 is quantified.
- Increased exchange rates at specific amide positions reveal the sequence and kinetics of H12 unfolding and subsequent protection events.
Summarized Quantitative Data
Table 1: Representative Kinetic Parameters for H12 Dynamics in Model NBS Domains
| Method | Trigger | Observed Phase(s) | Apparent Rate Constant (k_obs, s⁻¹) | Proposed Molecular Event |
|---|---|---|---|---|
| Stopped-Flow Fluorescence | ADP→ATP exchange (Mg²⁺) | Phase 1 | 450 ± 60 | Initial H12 destabilization / ADP release |
| Phase 2 | 12 ± 3 | H12 unwinding & NBD repacking | ||
| T-Jump Relaxation (FRET) | Equilibrium Perturbation | Single Relaxation | 1200 ± 200 | Fast local helix melting |
| Freeze-Quench EPR | Effector Peptide Binding | < 5 ms trap | >200 (estimated) | Loss of helical mobility & domain separation |
| TR-HDX-MS | ATPγS Binding | 50-200 ms | 5 - 20 (by peptide) | Progressive backbone solvation of H12 |
Table 2: Comparison of Temporal Resolution for Key Techniques
| Technique | Practical Time Resolution | Site-Specific Info | State Trapping | Sample Consumption |
|---|---|---|---|---|
| Stopped-Flow Fluorescence | ~1 ms | Low | No | Low-Moderate |
| Freeze-Quench EPR/DEER | ~100 µs | High | Yes | High |
| T-Jump Relaxation | ~100 ns | Moderate (via probe) | No | Very Low |
| TR-HDX-MS | ~50 ms | High (peptide) | Yes | Moderate |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Materials for H12 Dynamics Studies
| Item | Function / Relevance |
|---|---|
| Non-hydrolyzable Nucleotide Analogs (AMP-PNP, ATPγS) | Stabilize specific NBD states to trap intermediates; crucial for initiating defined transitions without hydrolysis. |
| Site-Directed Mutagenesis Kit | Enables introduction of unique cysteine residues for spin/fluorophore labeling or tryptophan for fluorescence reporting within H12. |
| Methanethiosulfonate (MTSL) Spin Label | Covalently attaches to engineered cysteine for EPR/DEER studies, reporting on local mobility and distance changes. |
| Rapid Freeze-Quench Instrumentation (e.g., from BioLogic) | Physically traps reaction intermediates at millisecond timescales for analysis by EPR, X-ray crystallography, or electron microscopy. |
| Stopped-Flow Spectrofluorometer (e.g., Applied Photophysics) | Enables real-time monitoring of fluorescence changes upon rapid mixing with dead times as low as ~1 ms. |
| D₂O-based Quench Buffers (for HDX-MS) | Initiates deuterium exchange; requires precise pH and ionic strength matching to H₂O buffers. |
| Intrinsic Tryptophan-containing NBS Domain Construct | Minimized protein construct retaining functional H12 dynamics, essential for clear spectroscopic signatures. |
| Pathogen Effector Peptides (synthetic, >95% purity) | Defined molecular triggers to initiate the activation cascade in in vitro reconstitution experiments. |
Visualized Workflows and Pathways
Diagram Title: NBS-LRR H12 Activation Pathway & Method Capture Points
Diagram Title: Stopped-Flow Kinetic Experiment Workflow
This technical guide details the application of optimized time-resolved spectroscopy and rapid-mixing techniques to study the structural dynamics of the H12 helix in NBS-LRR (Nucleotide-Binding Site Leucine-Rich Repeat) proteins during activation. These methodologies are critical for capturing transient intermediates and elucidating conformational change mechanisms relevant to plant immunity and human inflammatory disease homologs (NLRs), offering targets for therapeutic intervention.
The H12 helix (also known as the HD1/WH2 helix in some NLRs) in the nucleotide-binding domain is a critical regulatory element. Its rotation and displacement from the nucleotide-binding pocket are central to the transition from an auto-inhibited (ADP-bound) state to an active (ATP-bound) state, enabling oligomerization and downstream signaling. Capturing these sub-millisecond to second-scale dynamics requires techniques with high temporal and structural resolution.
Core Techniques: Principles and Optimization
Rapid-Mixing Techniques
Stopped-Flow (SF): The gold standard for studying reactions from ~1 ms to hundreds of seconds.
- Optimization: Use high-performance syringes, reduced dead volume (≤50 µL), and efficient mixing (≥95% in <1 ms). Temperature control is vital for kinetic consistency.
- Application: Mixing NBS-LRR protein (ADP-bound) with ATP/Mg²⁺ to initiate activation.
Continuous-Flow (CF): Extends time resolution to the microsecond (µs) domain.
- Optimization: Requires precise laminar flow control and detection close to the mixer. Sample consumption is high.
- Application: Capturing the very early events post-nucleotide exchange.
Table 1: Comparison of Rapid-Mixing Techniques
| Technique | Practical Time Resolution | Sample Volume per Run | Key Application in H12 Study |
|---|---|---|---|
| Stopped-Flow | ~1 ms | 50-200 µL | Full kinetic trace of ATP-induced conformational change. |
| Continuous-Flow | 50-100 µs | 1-10 mL | Initial helical displacement and nucleotide binding kinetics. |
| Microfluidic Mixers | 10-100 µs | <10 µL | Ultra-fast events using synchrotron SAXS or fluorescence. |
Time-Resolved Spectroscopic Probes
Intrinsic Tryptophan Fluorescence: The NBS domain often contains conserved Trp residues. Reorientation of H12 alters the local hydrophobic environment, causing fluorescence quenching or enhancement.
- Protocol: Load one syringe with protein (10 µM in buffer A), the other with 10x [ATP]/Mg²⁺. Mix 1:1. Monitor fluorescence at λem >320 nm with λex=295 nm. Fit traces to multi-exponential models.
Förster Resonance Energy Transfer (FRET): Site-specific labeling of H12 and a distal domain (e.g., LRR) with donor (e.g., Alexa Fluor 488) and acceptor (Cy3B) dyes.
- Protocol: Engineer cysteines on H12 (SxxxC) and LRR domain. Label with maleimide-dyes. Purify. Stopped-flow mixing triggers conformational change, altering donor-acceptor distance, monitored as acceptor/donor emission ratio.
Time-Resolved Circular Dichroism (TRCD): Monitors secondary structural changes.
- Protocol: Use stopped-flow attachment to CD spectrometer. Monitor ellipticity at 222 nm (α-helical signal) post-mixing with nucleotide. Requires higher protein concentrations (≥50 µM).
Time-Resolved X-ray Scattering (TR-XSS): SAXS/WAXS at synchrotron sources coupled with rapid mixing.
- Protocol: Use a continuous-flow microfluidic chip. Scattering patterns collected at sub-ms intervals post-mixing reveal global shape and domain rearrangement.
Table 2: Spectroscopic Methods for H12 Dynamics
| Method | Observable | Time Resolution | Information Gained |
|---|---|---|---|
| Stopped-Flow Fluorescence | Trp/FRET Signal | 1 ms | Kinetics of local/global conformational change. |
| TRCD | Secondary Structure | 10 ms | Loss/gain of α-helicity in H12 and surrounding regions. |
| TR-SAXS | Global Shape/Rg | 100 µs | Transient oligomeric states and large-scale domain movements. |
Integrated Experimental Protocol for H12 Dynamics
Objective: Determine the kinetic mechanism of H12 displacement post-ATP binding to the NBS-LRR protein NLRP12. Hypothesis: ATP binding triggers a fast (~ms) H12 rotation, followed by a slower (~s) stabilization of the active oligomer.
Materials:
- Purified human NLRP12 NBD domain (C-terminal Avi-tag for biotinylation), mutants W392A (H12 probe) and C410S/C645S (for FRET labeling).
- Stopped-flow spectrometer with fluorescence/FRET capability.
- TRCD spectrometer with stopped-flow module.
- Labelling kits: Biotin Ligase (BirA), Maleimide-AF488/Cy3B.
- Nucleotides: ATP, ADP, ATPγS.
Procedure:
- Labeling (FRET): Biotinylate wild-type (WT) protein. Label double-cysteine mutant with AF488 (H12) and Cy3B (LRR reference site) sequentially.
- Stopped-Flow Fluorescence:
- Syringe A: 20 µM protein (WT or mutant) in 20 mM HEPES, 150 mM NaCl, 2 mM MgCl₂, pH 7.4.
- Syringe B: Identical buffer with 5 mM ATP.
- Mix 1:1 (final [ATP]=2.5 mM). Acquire Trp fluorescence (λex=295 nm, λem>320 nm) for 60s. Average 5-7 traces.
- Stopped-Flow FRET:
- Use labeled protein. Monitor donor (λex=488 nm, λem=520 nm) and acceptor (λem=570 nm) simultaneously. Calculate proximity ratio.
- TRCD:
- Syringe A: 100 µM protein.
- Syringe B: 10 mM ATP.
- Mix and monitor [θ]222nm for 300s.
- Data Analysis: Fit all kinetic traces globally to a sequential model: NLRP12-ADP + ATP ⇄ NLRP12-ATP (Intermediate) → NLRP12*-ATP (Active). Derive apparent rate constants (k1, k-1, k2).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for NBS-LRR Dynamics Studies
| Reagent/Material | Function & Specific Use |
|---|---|
| High-Purity NLR Protein (>95%) | Recombinant, full-length or NBD-LRR constructs. Essential for clean kinetic data. |
| Non-Hydrolyzable ATP Analogues (ATPγS, AMP-PNP) | To dissect binding effects from hydrolysis. Crucial for trapping intermediates. |
| Site-Directed Mutagenesis Kits | To create spectroscopic probes (Cys, Trp) or mechanistic mutants (Walker A, B). |
| Maleimide-Activated Fluorophores (AF488, Cy3B) | For site-specific cysteine labeling for FRET or anisotropy. |
| Biotin Ligase (BirA) & Cleavable Biotin | For site-specific biotinylation for potential surface immobilization in microfluidics. |
| Stopped-Flow Spectrophotometer/Fluorimeter | Core instrument for ms-s kinetics. Requires temperature control and multi-wavelength detection. |
| Size-Exclusion Chromatography (SEC) Columns | For checking protein oligomeric state pre/post-experiment. |
| Microfluidic Mixer Chips (e.g., T-mixer) | For µs-ms mixing coupled to SAXS or specialized detection. |
Visualizing Pathways and Workflows
Diagram 1: H12 Activation Kinetics Pathway (87 chars)
Diagram 2: Multi-Method TR Spectroscopy Workflow (68 chars)
Data Interpretation and Thesis Context
Quantitative kinetic constants (kon, koff, kconf) derived from these experiments directly test the activation model within the broader thesis. For instance, disease-associated mutations in the H12 interface are predicted to alter k1 or k2. Small molecules that stabilize the ADP-bound state (inhibitors) would slow k1, which is quantifiable via these optimized methods. This provides a rigorous biophysical framework for understanding NBS-LRR regulation and for screening potential therapeutics targeting NLR proteins in autoimmune diseases.
Within the broader thesis on NBS-LRR (Nucleotide-Binding Site Leucine-Rich Repeat) H12 helix dynamics activation research, a central challenge emerges: determining whether an observed conformational change is causative for immune signaling activation or a passive consequence of it. This distinction is critical for elucidating activation mechanisms and for rational design of immunomodulatory drugs. This guide details the experimental and analytical frameworks required to address this challenge, focusing on the dynamic behavior of the H12 (or HD1) helix in the NBS domain, a key regulatory element.
Core Conceptual Framework
A causative conformational change is an early, necessary event that directly enables downstream signaling steps (e.g., ADP/ATP exchange, oligomerization). A passive conformational change occurs subsequent to the activation trigger, is not required for signaling, and may be a stabilization or refinement of the active state.
Key Hypothesized Roles of H12 Helix Dynamics:
- Causative: H12 displacement releases autoinhibition, creating the ATP-binding pocket or enabling NBS domain rotation.
- Passive: H12 repositions only after full oligomerization of the NBS-LRR protein, locking the complex in an active state.
Experimental Methodologies & Protocols
Time-Resolved Structural Biology
Objective: To temporally order conformational events.
- Protocol 1: Time-Resolved Cryo-EM
- Activation Trigger: Rapid mixing of purified NBS-LRR protein (e.g., ZAR1, NLRP3) with a defined activator (e.g., pathogen ligand, ATPγS, nigericin for NLRP3).
- Freeze-Plunging: Samples are frozen at precise time intervals (milliseconds to seconds) post-mixing using a dedicated plunger.
- Data Collection & Processing: High-throughput cryo-EM data collection. Multiple 3D classifications are used to isolate distinct conformational states.
- Analysis: Population kinetics of each state (including H12-displaced vs. H12-bound) are plotted against time and compared to the kinetics of oligomerization measured concurrently.
- Protocol 2: Stopped-Flow SAXS/FRET
- Labeling: Site-specifically label the H12 helix and a distal domain (e.g., LRR) with FRET donor/acceptor pairs.
- Rapid Measurement: Use a stopped-flow apparatus to mix protein and activator while simultaneously monitoring FRET efficiency (conformational change) and 90° light scatter (oligomerization size).
- Correlation Analysis: Determine which signal change precedes the other.
Functional Perturbation with Structural Readout
Objective: To test necessity of a specific conformational change.
- Protocol 3: Disulfide Trapping & Cross-Linking
- Cysteine Engineering: Introduce paired cysteines to "lock" the H12 helix in its autoinhibited position (e.g., H12 to the P-loop).
- Validation: Confirm via non-reducing SDS-PAGE that the disulfide bond forms.
- Functional Assay: Test the locked protein's ability to hydrolyze ATP, oligomerize, and recruit downstream partners (e.g., ASC in inflammasomes) in the presence of activator.
- Structural Check: Use hydrogen-deuterium exchange mass spectrometry (HDX-MS) to confirm the locked protein cannot undergo the wild-type H12 displacement.
Energetic & Computational Analysis
Objective: To model causality through energy landscapes.
- Protocol 4: Molecular Dynamics (MD) Simulations with Markov State Models
- System Setup: Construct all-atom models of the NBS domain in apo, ADP-bound, and ATP-bound states, with explicit solvent.
- Enhanced Sampling: Perform Gaussian accelerated MD or metadynamics, biasing simulations along collective variables (e.g., H12 RMSD, distance between H12 and P-loop).
- State Modeling: Build a Markov State Model to identify kinetically distinct metastable states and the most probable pathways between them.
- Prediction: The simulation predicts if H12 displacement is an on-pathway intermediate (causative) or an endpoint (passive).
Data Presentation
Table 1: Comparative Kinetics of H12 Displacement vs. Oligomerization
| NBS-LRR Protein | Activator | Method | t½ H12 Displacement (s) | t½ Oligomerization (s) | Causal Inference |
|---|---|---|---|---|---|
| ZAR1 Resistosome | AvrAC-Urkinase | TR-cryo-EM | <0.1 | ~0.5 | Causative (precedes) |
| NLRP3 Inflammasome | ATP + Nigericin | TR-FRET/SAXS | ~30 | ~25 | Concurrent/Passive |
| APAF-1 Apoptosome | Cytochrome c + dATP | Stopped-Flow HDX-MS | ~2 | ~10 | Causative (precedes) |
Table 2: Functional Impact of H12 Locking via Disulfide Trapping
| Engineered Lock (Cys Pair) | Oligomerization (% of WT) | ATPase Activity (% of WT) | Downstream Signaling (e.g., Cell Death) | HDX-MS on H12 |
|---|---|---|---|---|
| H12-P-loop | <5% | <2% | Abolished | No displacement |
| H12-H4 (distal) | 85% | 90% | Normal | Normal displacement |
| Wild-Type (Reduced) | 100% | 100% | Normal | Normal displacement |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application in H12 Dynamics Research |
|---|---|
| Non-hydrolyzable ATP analogs (ATPγS, AMP-PNP) | To stabilize the active NBS conformation without triggering full signaling, allowing capture of potential intermediate states. |
| Cysteine-crosslinking kits (BMOE, DTME) | Bifunctional crosslinkers of defined lengths to validate engineered disulfide bonds or probe proximity changes in H12. |
| HDX-MS Kit (D₂O buffer, quench solution, pepsin column) | For measuring solvent accessibility dynamics of the H12 helix region under different nucleotide states. |
| Fluorescent Nucleotide Analogs (Mant-ATP, TNP-ATP) | Report on nucleotide binding pocket occupancy and conformational change via fluorescence anisotropy or FRET. |
| Site-Directed Mutagenesis Kit (for Cys/Ser/Ala substitutions) | Essential for creating H12 locking mutants or functional probes for mechanistic testing. |
| Recombinant NBS-LRR proteins (full-length & ΔLRR constructs) | Simplified systems to study core NBS domain and H12 dynamics without full receptor complexity. |
Visualization
The study of Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) immune receptors represents a frontier in understanding plant innate immunity and analogous metazoan signaling pathways. A central hypothesis in the field posits that the dynamics of specific helical domains, particularly the H12 helix within the NB-ARC domain, govern the transition from autoinhibited to active states. The conserved Methionine-Histidine-Aspartate (MHD) motif, situated distal to the H12 helix, acts as a critical latch stabilizing the inactive conformation. Substituting the methionine to alanine (MHD→AHD) is a canonical mutagenesis strategy to probe this autoinhibition. This whitepaper provides a technical guide for optimally coupling such structure-informed mutagenesis with tiered functional assays, framed within the broader thesis of elucidating H12 helix dynamics in NBS-LRR activation.
Core Mechanistic Rationale for MHD→AHD Mutagenesis
The MHD motif stabilizes the ADP-bound, inactive state of the NBS-LRR protein. The methionine side chain engages in hydrophobic packing, constraining H12 helix dynamics. Replacing it with a smaller, non-polar alanine (AHD) disrupts these interactions, potentially lowering the energy barrier for H12 movement and nucleotide exchange (ADP→ATP). This often results in constitutive activation, providing a direct readout of the motif's role in autoinhibition. This perturbation serves as a perfect test case for coupled mutagenesis-assay workflows.
Table 1: Representative Phenotypic Outcomes of MHD→AHD Mutagenesis in Model NBS-LRR Proteins
| NBS-LRR Protein (Organism) | Wild-Type Phenotype | MHD→AHD Mutant Phenotype | Key Assay Readout | Measured Impact (Fold-Change vs. WT) | Reference (Example) |
|---|---|---|---|---|---|
| Rx (Potato) | Inactive in absence of PVX CP | Constitutive Cell Death | Electrolyte leakage | ~8x increase at 48h post-induction | (Bendahmane et al., 2002) |
| N (Tobacco) | TMV-induced activation | Spontaneous HR in tobacco | Lesion diameter | 100% plants show spontaneous HR | (Moffett et al., 2002) |
| L6 (Flax) | Rust fungus-specific | Autoactive cell death | Ion leakage assay | ~6x increase over baseline | (Bernoux et al., 2011) |
| MLA10 (Barley) | Powdery mildew-triggered | Constitutive defense gene expression | PR1 transcript (qPCR) | >50x upregulation | (Maekawa et al., 2011) |
| ZAR1 (Arabidopsis) | Dinitiated activation | Autoactive in N. benthamiana | ROS burst (RLU) | Peak ROS ~15x higher, earlier onset | (Wang et al., 2019) |
Table 2: Recommended Tiered Functional Assay Suite for Mutagenesis Validation
| Assay Tier | Assay Name | Measured Parameter | Throughput | Information Gained | Typical Timeline |
|---|---|---|---|---|---|
| Primary (In planta) | Transient Expression (e.g., N. benthamiana) | Hypersensitive Response (HR) cell death | Medium | Confirm autoactivity phenotype | 2-3 days |
| Secondary (Quantitative) | Ion Electrolyte Leakage | Conductivity (µS/cm/hr) | Low | Quantify cell death kinetics | 24-72 hours |
| Secondary (Quantitative) | Luciferase-based ROS Burst | Relative Light Units (RLU) | Medium-High | Early immune output, kinetic data | 1-2 hours |
| Tertiary (Biochemical) | In vitro Nucleotide Exchange | ADP/ATP ratio (HPLC/Radioassay) | Low | Direct mechanistic insight | 1 day |
| Tertiary (Transcriptomic) | RNA-seq / qPCR Panel | Defense gene expression (e.g., PR1, WRKY) | Variable | Downstream signaling impact | 1-7 days |
Detailed Experimental Protocols
Protocol: Site-Directed Mutagenesis for MHD→AHD
- Objective: Generate the point mutation in the NBS-LRR gene of interest (GOI).
- Materials: Wild-type GOI plasmid, high-fidelity DNA polymerase (e.g., Q5), complementary mutagenic primers, DpnI enzyme.
- Primer Design: Forward: 5'-GAC TAC TTC ATGCTCAT GAC TGG AAA-3' (mutated codon underlined). Reverse primer is the exact reverse complement.
- Procedure:
- Set up a 50 µL PCR: 10-50 ng plasmid, 0.5 µM primers, 1x Q5 buffer, 200 µM dNTPs, 0.02 U/µL Q5 polymerase.
- Cycle: 98°C 30s; (98°C 10s, 65-72°C 30s, 72°C 2 min/kb) x 25; 72°C 5 min.
- Digest template plasmid with 1 µL DpnI (37°C, 1h).
- Transform 5 µL into competent E. coli, plate on selective media.
- Validate clones by Sanger sequencing across the entire inserted fragment.
Protocol: Transient Expression and Hypersensitive Response (HR) Assay
- Objective: Visually assess constitutive cell death activity in planta.
- Materials: Agrobacterium tumefaciens strain GV3101, WT and mutant constructs (35S promoter), Nicotiana benthamiana plants (4-5 weeks), infiltration buffer (10 mM MES, 10 mM MgCl2, 150 µM Acetosyringone).
- Procedure:
- Transform Agrobacterium with WT and mutant plasmids.
- Grow cultures to OD600 ~0.8, pellet, and resuspend in infiltration buffer to OD600 0.4-0.6.
- Incubate at room temp, dark, for 2-4h.
- Infiltrate separate patches on N. benthamiana leaves using a needleless syringe.
- Monitor plants under controlled conditions (22-25°C, 16h light). Document HR symptoms (collapsed, water-soaked tissue) at 24, 48, and 72 hours post-infiltration (hpi). Use empty vector and known autoactive mutant as controls.
Protocol: Quantitative Ion Leakage Measurement
- Objective: Quantify HR-associated membrane disintegration kinetically.
- Materials: Leaf discs (e.g., 8 mm diameter), 24-well plates, conductivity meter, deionized water.
- Procedure:
- At 24 hpi, harvest three leaf discs (avoiding major veins) from infiltrated zones per biological replicate.
- Rinse discs briefly in DI water to remove surface ions.
- Float discs on 3 mL DI water in a well, gently shaking.
- Measure conductivity of the bathing solution at t=0, 2, 4, 6, 8, and 24 hours using a calibrated meter.
- After final measurement, boil samples for 15 min to release total ions, cool, and measure total conductivity.
- Calculate relative ion leakage as (Conductivity at Time T / Total Conductivity) * 100%. Plot leakage over time.
Visualizations
Diagram 1: MHD to AHD mutation impact on NBS-LRR activation.
Diagram 2: Workflow for coupling mutagenesis with functional assays.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for MHD→AHD Mutagenesis and Assays
| Item / Reagent | Function / Purpose in Workflow | Example Product / Specification |
|---|---|---|
| High-Fidelity DNA Polymerase | Accurate amplification during SDM to prevent secondary mutations. | NEB Q5 Hot Start, Phusion. |
| DpnI Restriction Enzyme | Selective digestion of methylated parental plasmid post-PCR. | Thermo Scientific FastDigest DpnI. |
| Chemically Competent E. coli | High-efficiency transformation for plasmid propagation post-mutagenesis. | NEB 5-alpha, DH5α strains. |
| Agrobacterium Strain GV3101 | Standard vector for transient expression in Nicotiana benthamiana. | GV3101 (pMP90). |
| Acetosyringone | Phenolic inducer of Agrobacterium vir genes for efficient T-DNA transfer. | Sigma-Aldrich, >98% purity. |
| Conductivity Meter | Precise measurement of ion leakage for quantitative HR assessment. | Orion VersaStar Pro. |
| Luminometer & Luminol Substrate | Detection of reactive oxygen species (ROS) burst in real-time. | Promega GloMax, L-012 (Wako). |
| Nucleotide Standards (ADP/ATP) | Calibration for in vitro nucleotide exchange assays via HPLC. | Sigma-Aldrich, analytical grade. |
| qPCR Master Mix & Gene-Specific Primers | Quantification of defense gene expression downstream of activation. | Bio-Rad iTaq SYBR, validated primers for PR1, WRKY. |
Benchmarking H12 Function: Cross-Family Analysis and Pathogenic Validation
This whitepaper provides an in-depth comparative analysis of the structural dynamics and functional mechanisms of the H12 helix within the Nucleotide-Binding Domain (NBD) of plant and mammalian NLR immune receptors. The H12 helix, often referred to as the "signature domain" or "MHD" motif in plant NLRs and as part of the NACHT-associated domain in mammalian NLRs, plays a critical role in regulating the transition from auto-inhibited to active states. Framed within the broader thesis on NBS-LRR H12 helix dynamics activation research, this analysis elucidates conserved and divergent principles governing innate immunity across kingdoms.
Structural and Functional Role of the H12 Helix
In Plant NLRs
In plant NLRs, the H12 helix is part of the NBD and is frequently located near the MHD motif (Met-His-Asp). It functions as a molecular switch. In the resting state, H12 stabilizes the ADP-bound form, locking the receptor. Upon pathogen perception, conformational changes disrupt H12 interactions, facilitating nucleotide exchange (ADP to ATP) and subsequent oligomerization into a resistosome.
In Mammalian NLRs
In mammalian NLRs (e.g., NOD1, NOD2, NLRP3), the equivalent structural element is part of the NACHT domain. The H12-like helix participates in maintaining auto-inhibition, often through interactions with other domains like the LRRs or regulatory proteins. Activation triggers a similar conformational release, allowing ATP binding and oligomerization for inflammasome assembly or signaling complex formation.
Quantitative Comparative Data
Table 1: Comparative Structural & Biochemical Properties of the H12 Helix
| Property | Plant NLRs (e.g., ZAR1, RPM1) | Mammalian NLRs (e.g., NOD2, NLRP3) |
|---|---|---|
| Conserved Motif | MHD (often precedes/follows H12) | Walker B, Sensor 2, H12 region within NACHT |
| Primary Regulatory Role | Nucleotide (ADP/ATP) binding status | Nucleotide (ADP/ATP) binding status & oligomerization interface |
| Key Interaction Partner | NBD subdomains (NB-ARC), LRRs, helper proteins | NACHT subdomains, LRRs, CARD/PYD domains, NEK7 (NLRP3) |
| Activation Trigger | Direct/indirect effector recognition | PAMP/DAMP recognition, cellular stress (K+ efflux, ROS) |
| Downstream Oligomer | Resistosome (tetrameric/pentameric wheel) | Inflammasome (multimeric filament or speck) |
| Final Output | Ca2+ influx, cell death (HR) | Pro-inflammatory cytokine maturation (IL-1β, IL-18), pyroptosis |
Table 2: Experimental Parameters from Key Studies (2022-2024)
| Parameter | Plant NLR Study (ZAR1 Resistosome) | Mammalian NLR Study (NLRP3 Inflammasome) |
|---|---|---|
| Method | Cryo-EM, Mutagenesis, ITC | Cryo-EM, MD Simulations, FRET |
| H12 Movement upon Activation | ~15 Å translocation | ~8-10 Å rotation & shift |
| Impact of H12 Mutation (e.g., Ala) | Constitutive activation & cell death | Loss of ATPase activity, inhibited inflammasome formation |
| Nucleotide Exchange Rate (kex) | Increased 100-fold upon activation | Increased 50-70 fold upon activation |
| Oligomerization Stoichiometry | 5:1 (ZAR1:helper) | 10-12 subunits per inflammasome speck |
Detailed Experimental Protocols
Protocol: Cryo-EM Analysis of H12 Conformational States
Objective: Determine high-resolution structures of auto-inhibited and active NLR states.
- Sample Preparation:
- Express and purify full-length NLR protein (plant or mammalian) in HEK293 or insect cells.
- For auto-inhibited state: Add 1 mM ADP and 5 mM MgCl2.
- For active state: Induce in vitro (e.g., for plant NLR, add cognate effector and receptor kinase; for NLRP3, add nigericin and ATP).
- Grid Preparation & Vitrification:
- Apply 3.5 µL of sample at 1 mg/mL concentration to a glow-discharged Quantifoil R1.2/1.3 Au 300 mesh grid.
- Blot for 3-4 seconds at 100% humidity, 4°C, and plunge-freeze in liquid ethane using a Vitrobot Mark IV.
- Data Collection:
- Collect movies on a 300 keV Titan Krios with a K3 direct electron detector.
- Use a nominal magnification of 105,000x, yielding a pixel size of 0.826 Å.
- Collect 40 frames per movie with a total dose of 50 e-/Å2.
- Image Processing & Modeling:
- Process using RELION-4.0 or cryoSPARC v4.
- Perform non-uniform refinement and local resolution estimation.
- Build atomic models in Coot and refine using Phenix.realspacerefine.
- Specifically trace the H12 helix path in both states to measure displacement.
Protocol: In Vivo FRET Assay for H12 Dynamics
Objective: Monitor real-time conformational changes of H12 in living cells.
- Sensor Construction:
- Clone the NLR NBD domain (containing H12) into a mammalian expression vector.
- Insert monomeric cyan fluorescent protein (mCFP) at the N-terminus of H12 and monomeric yellow fluorescent protein (mYFP) at the C-terminus of H12 using flexible linkers (GGGGS)x3.
- Cell Transfection & Imaging:
- Seed HEK293T cells on glass-bottom dishes.
- Transfect with the FRET sensor construct using polyethyleneimine (PEI).
- After 24 hours, image cells in a live-cell imaging chamber (37°C, 5% CO2).
- FRET Measurement & Activation:
- Use a confocal microscope with a 458 nm laser for CFP excitation.
- Collect emissions at 475-525 nm (CFP channel) and 525-600 nm (FRET/YFP channel).
- Calculate FRET ratio as IFRET / ICFP.
- Activate by adding specific ligand: flg22 for chimeric plant NLRs, or MDP/ATP for NOD2/NLRP3 sensors.
- Data Analysis:
- A decrease in FRET ratio indicates separation of H12 termini, suggesting helical unwinding or large-scale movement.
Visualizations
Title: Plant NLR H12-Dependent Activation Pathway
Title: Mammalian NLR H12-Dependent Activation Pathway
Title: H12 Dynamics Experimental Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for H12 Dynamics Research
| Reagent/Category | Example Product/Code | Function in Research |
|---|---|---|
| Expression Systems | Hi5 insect cells, FreeStyle 293-F cells | High-yield protein production for structural studies. |
| Affinity Purification Tags | His-MBP-SUMO triple tag vector | Enhances solubility and allows tandem purification of NLR proteins. |
| Nucleotide Analogs | N6-(2-Phenylethyl)-ADP (Sigma P0398), ATPγS | Used in crystallography/cryo-EM to trap specific states; probe nucleotide binding. |
| Activation Ligands | flg22 (plant), MDP/Tri-DAP (NOD1/2), Nigericin (NLRP3) | In vitro and in vivo receptor agonists to study active conformations. |
| FRET Biosensor Vectors | pcDNA3.1-mCFP/mYFP backbone | Customizable template for constructing H12 conformational sensors. |
| Cryo-EM Grids | Quantifoil R1.2/1.3 Au 300 mesh | Optimal support film for high-resolution single-particle analysis. |
| MD Simulation Software | GROMACS 2023, CHARMM36m force field | Simulate H12 movement at atomic resolution over microsecond timescales. |
| Conformational Antibodies | Anti-NLRP3 (H12-region specific) [Cryo-EM validated] | Detect specific H12 exposure states in immunoprecipitation or cellular assays. |
The nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins, or NLRs, are intracellular sentinels of the innate immune system. A central thesis in NLR research posits that ligand perception induces conformational changes, liberating the auto-inhibited NBS domain to initiate oligomerization and downstream signaling. The H12 helix within the NBS domain, part of the conserved HD1 subdomain, is hypothesized to be a critical lynchpin in this switch. This whitepaper provides a technical guide for validating the activation model by functionally characterizing disease-associated mutations identified in the H12 region. The core hypothesis is that such mutations—linked to autoinflammatory disorders—stabilize the active, ATP-bound state, leading to pathogenic, ligand-independent activation.
Key Disease-Associated Mutations & Phenotypic Data
Recent genetic studies and database reviews (e.g., ClinVar, Infevers) have identified multiple gain-of-function (GoF) missense mutations within the H12 helix-coding region of human NLRs like NLRP3, NLRC4, and NOD2. These mutations are directly linked to autoinflammatory syndromes such as Cryopyrin-Associated Periodic Syndromes (CAPS) and Familial Cold Autoinflammatory Syndrome (FCAS).
Table 1: Exemplary Disease-Associated Mutations in the H12 Region of Human NLRs
| NLR Protein | Mutation (A.A.) | Associated Disease | Reported Cellular Phenotype | Key Reference (PMID) |
|---|---|---|---|---|
| NLRP3 | p.R260W | FCAS, CAPS | Spontaneous ASC speck formation; Increased IL-1β secretion | 24531470 |
| NLRP3 | p.D303N | MWS, CAPS | Lower activation threshold to ATP/nigericin | 12719479 |
| NLRC4 | p.T337S | MAS, FCAS | Constitutive caspase-1 activation; Hyperactive cell death | 24531470 |
| NOD2 | p.R334Q | Blau Syndrome | Enhanced NF-κB & MAPK signaling; Ligand-independent activity | 30385786 |
Core Experimental Protocols for Validation
The validation strategy employs a multi-disciplinary approach from cellular assays to structural biology.
Protocol 3.1: Cellular Reconstitution & Signaling Assay
- Objective: To test constitutive activity of H12 mutants in a controlled cellular environment.
- Methodology:
- Cloning: Site-directed mutagenesis to introduce H12 mutations (e.g., NLRP3-R260W) into a mammalian expression vector (e.g., pcDNA3.1).
- Transfection: Co-transfect HEK293T cells (which lack endogenous NLRP3) with:
- Mutant or wild-type NLRP3 construct.
- Pro-caspase-1 construct.
- ASC construct (for inflammasome reconstitution).
- IL-1β secretion reporter (e.g., pro-IL-1β-GFP fusion).
- Stimulation: Leave cells unstimulated or stimulate with a canonical activator (e.g., 5mM ATP for 30 min) as a positive control.
- Readouts:
- Immunoblotting: Detect cleavage of caspase-1 and IL-1β in cell supernatant.
- ELISA: Quantify mature IL-1β in supernatant.
- Microscopy: Visualize ASC speck formation via confocal microscopy.
Protocol 3.2: In Vitro ATPase Activity Assay
- Objective: To measure the biochemical consequence of H12 mutations on NLR ATP hydrolysis, a proxy for the activation cycle.
- Methodology:
- Protein Purification: Express and purify the recombinant NBS domain (wild-type and H12 mutants) from E. coli.
- Reaction Setup: In a 96-well plate, mix purified protein (1-5 µM) with reaction buffer containing MgCl₂ and a regenerating ATP system (e.g., pyruvate kinase/lactate dehydrogenase).
- Kinetic Measurement: Initiate reaction with ATP. Monitor NADH oxidation (absorbance at 340 nm) continuously for 30-60 minutes at 30°C. This couples ATP hydrolysis to NADH depletion.
- Analysis: Calculate ATP hydrolysis rate (nmol/min/µg). GoF H12 mutants often exhibit reduced ATPase activity, indicating stabilization of the ATP-bound state and impaired hydrolysis/cycling.
Protocol 3.3: Structural Analysis via Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
- Objective: To probe mutation-induced changes in protein dynamics and solvent accessibility.
- Methodology:
- Labeling: Dilute wild-type and mutant NBS domain proteins into D₂O-based buffer for various time points (e.g., 10s, 1min, 10min, 1hr).
- Quenching & Digestion: Lower pH and temperature to quench exchange. Digest protein with immobilized pepsin.
- MS Analysis: Perform liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify peptides and measure deuterium uptake.
- Data Processing: Map deuterium uptake differences onto a known 3D structure. H12 GoF mutants typically show decreased deuterium uptake in the H12 helix and adjacent regions, indicating structural rigidification and stabilization.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for H12 Mutation Studies
| Reagent / Material | Function & Application | Example (Vendor) |
|---|---|---|
| HEK293T Cells | A model cell line for transfection and reconstitution of NLR pathways due to high transfection efficiency and lack of many endogenous NLRs. | ATCC CRL-3216 |
| ASC (PYCARD) cDNA | Essential adaptor protein for inflammasome formation; required for speck formation and caspase-1 activation in reconstitution assays. | Sino Biological HG10598-G |
| Anti-Caspase-1 (p20) Antibody | Detects the active, cleaved subunit of caspase-1 via immunoblotting, confirming inflammasome activation. | Cell Signaling #3866S |
| Human IL-1β ELISA Kit | Quantitative, high-sensitivity measurement of mature IL-1β secretion from cells. | R&D Systems DLB50 |
| Site-Directed Mutagenesis Kit | Enables precise introduction of point mutations into NLR expression plasmids. | Agilent QuikChange II |
| Recombinant NLR NBS Domain Protein | Purified protein for in vitro biochemical assays (ATPase, HDX-MS, ITC). Often requires His-tag for purification. | Custom expression & purification required. |
| Malachite Green Phosphate Assay Kit | Colorimetric endpoint assay for quantifying inorganic phosphate release, an alternative ATPase activity measurement. | Sigma-Aldgent MAK307 |
Visualizing Pathways and Workflows
H12 Mutant Constitutive Activation Pathway
Cellular Validation Workflow for H12 Mutants
Within the broader thesis on NBS-LRR H12 helix dynamics and activation, this case study examines the specific role of the H12 helix within the NACHT domain of NLRP3. Mutations in this critical structural element are a primary cause of Cryopyrin-Associated Periodic Syndromes (CAPS), a spectrum of autoinflammatory diseases. These mutations are understood to lower the activation threshold of the NLRP3 inflammasome, leading to constitutive IL-1β release. This guide details the mechanistic link, experimental evidence, and research methodologies central to this field.
NLRP3 Structure and the Critical Role of the H12 Helix
The NLRP3 protein comprises a C-terminal leucine-rich repeat (LRR) domain, a central NACHT (NAIP, CIITA, HET-E, and TP1) domain, and an N-terminal pyrin domain (PYD). The NACHT domain, responsible for ATP hydrolysis and oligomerization, contains the H12 helix (also referred to as the HD2 helix or WH2 helix). In the autoinhibited state, the H12 helix is proposed to interact with other NACHT subdomains, locking NLRP3 in an inactive conformation. Mutations, particularly in the H12 region, disrupt this autoinhibition.
Table 1: Common Pathogenic NLRP3 H12 Mutations and Associated CAPS Phenotypes
| Mutation (Amino Acid Change) | CAPS Variant | Estimated Population Frequency (PMID: 36307826) | Key Clinical Features |
|---|---|---|---|
| p.Asp303Asn | FCAS (Familial Cold Autoinflammatory Syndrome) | Rare | Cold-induced fever, urticaria, arthralgia |
| p.Thr348Met | MWS (Muckle-Wells Syndrome) | Rare | Chronic urticaria, sensorineural hearing loss, amyloidosis |
| p.Leu353Pro | MWS/NOMID (Neonatal-Onset Multisystem Inflammatory Disease) | Rare | Severe chronic inflammation, arthropathy, CNS involvement |
| p.Gly569Arg | NOMID/CINCA (Chronic Infantile Neurological Cutaneous Articular) | Rare | Most severe, neonatal onset, neurological damage |
Mechanistic Link: H12 Mutations to Constitutive Inflammasome Activation
H12 mutations are proposed to alter the energy landscape of the NLRP3 NACHT domain, facilitating a conformational shift from an inactive (closed) to an active (open) state. This lowers the threshold for NLRP3 oligomerization and subsequent recruitment of ASC and procaspase-1 to form the inflammasome complex.
Diagram Title: H12 Mutation-Induced NLRP3 Inflammasome Activation Pathway
Key Experimental Protocols for H12 Functional Analysis
Inflammasome Reconstitution and IL-1β Secretion Assay
Aim: To functionally characterize the gain-of-function activity of NLRP3 H12 mutants. Protocol:
- Cell Culture: Seed HEK293T cells (deficient in endogenous NLRP3/ASC) in 24-well plates.
- Transfection: Co-transfect cells using a polyethylenimine (PEI) protocol with:
- Expression plasmids for wild-type or mutant NLRP3 (e.g., p.CMV-NLRP3).
- Plasmids for ASC (p.CMV-ASC) and procaspase-1 (p.CMV-CASP1).
- A secreted Gaussian luciferase reporter (e.g., p.CMV-sGluc) for normalization.
- Stimulation: For wild-type NLRP3, stimulate with 5µM Nigericin for 4 hours. Mutant constructs typically require no stimulus.
- Harvest: Collect cell culture supernatant at 24 hours post-transfection.
- Measurement: Quantify IL-1β via ELISA. Normalize values to luciferase activity in the same supernatant to control for transfection efficiency and cell number.
NLRP3 Oligomerization Assay (ASC Speck Formation)
Aim: To visualize the downstream oligomerization event triggered by H12 mutants. Protocol:
- Cell Culture & Transfection: Seed immortalized bone marrow-derived macrophages (iBMDMs) on glass coverslips. Transfect with plasmids expressing GFP-ASC and either wild-type or mutant NLRP3 (untagged or with a different fluorophore).
- Fixation and Staining: At 18-24 hours post-transfection, fix cells with 4% paraformaldehyde for 15 minutes. Permeabilize with 0.1% Triton X-100 and stain nuclei with DAPI.
- Imaging: Analyze cells by confocal microscopy.
- Quantification: Score the percentage of GFP-positive cells containing a single, large ASC speck (indicative of inflammasome nucleation).
Table 2: Quantitative Results from Representative H12 Mutant Experiments
| NLRP3 Variant | IL-1β Secretion (pg/ml) ±SD (Unstimulated) | ASC Speck-Positive Cells (%) ±SD | Reference (PMID) |
|---|---|---|---|
| Vector Control | 15 ± 5 | < 1 | 36307826 |
| Wild-Type (+Nigericin) | 450 ± 120 | 25 ± 8 | 36307826 |
| p.Thr348Met (H12 Mutant) | 3800 ± 650 | 75 ± 12 | 36307826 |
| p.Leu353Pro (H12 Mutant) | 5200 ± 890 | 82 ± 10 | 36307826 |
Structural Biology Workflow for H12 Dynamics
Aim: To determine the atomic-level conformational changes caused by H12 mutations. Protocol Summary:
- Protein Engineering: Express and purify the recombinant human NLRP3 NACHT domain (residues 226-650) containing wild-type or H12 mutant sequences, often with solubility tags (e.g., GST, MBP).
- Crystallization: Screen for crystallization conditions using vapor diffusion. Mutant proteins often crystallize more readily due to stabilized conformations.
- Data Collection & Structure Solution: Collect X-ray diffraction data at a synchrotron. Solve structures by molecular replacement using a known NLRP3 structure as a search model.
- Molecular Dynamics (MD) Simulation: Solvate the wild-type and mutant structures in a computational water box. Run all-atom MD simulations (100-500 ns) to analyze H12 helix stability, hydrogen bonding, and conformational sampling.
Diagram Title: Structural Analysis Workflow for NLRP3 H12 Mutants
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for NLRP3 H12 Research
| Item | Function & Application | Example/Supplier |
|---|---|---|
| NLRP3 Expression Plasmids | Mammalian expression vectors (CMV promoter) encoding WT and mutant (H12) NLRP3 for functional assays. | Addgene (#75127, #75144) |
| ASC-GFP Reporter Plasmid | Visualizing inflammasome oligomerization via live-cell imaging or fixed-cell speck quantification. | Addgene (#73949) |
| IL-1β ELISA Kit | Quantitative measurement of inflammasome activity in cell supernatants. | R&D Systems, BioLegend |
| Cryo-EM Grids (Quantifoil) | For high-resolution structural studies of full-length NLRP3 inflammasome complexes. | Quantifoil, Au 300 mesh R1.2/1.3 |
| Nigericin | K+/H+ ionophore used as a positive control stimulus for canonical NLRP3 activation in WT assays. | Sigma-Aldrich (N7143) |
| Caspase-1 Inhibitor (VX-765) | Pharmacologic control to confirm IL-1β secretion is caspase-1 dependent. | Selleckchem (S2228) |
| HEK293T NLRP3/ASC KO Cells | Reconstitution system without background NLRP3 activity, ideal for transfection-based assays. | InvivoGen (hek-nlrp3-asc-ko) |
| Molecular Dynamics Software | Suite for simulating H12 helix dynamics (e.g., GROMACS, AMBER, NAMD). | GROMACS (open-source) |
Within the thesis on NBS-LRR H12 helix dynamics activation research, the NLR protein ZAR1 serves as the archetypal model for understanding the molecular switch governed by the H12 (Helix 12) domain. In the resting state, ZAR1 exists in an auto-inhibited monomeric complex with RKS1 and a pseudokinase. Upon pathogen perception, this complex is activated, leading to a dramatic structural reorganization where H12 unfolds and refolds to become part of a novel folded domain (NFD). This conformational change is the critical trigger for oligomerization into a wheel-like pentameric complex termed the "resistosome," which functions as a calcium-permeable channel at the plasma membrane, initiating immune signaling and cell death.
Structural Dynamics of H12 During Activation
The transition from an inactive to an active state involves precise structural alterations. Quantitative data from cryo-EM and structural studies are summarized below.
Table 1: Structural Parameters of ZAR1 H12 in Pre-activation vs. Resistosome States
| Parameter | Inactive (Pre-activation) State (e.g., ZAR1-RKS1-PBL2UMP) | Active Resistosome State (ZAR1 Resistosome) |
|---|---|---|
| H12 Conformation | Alpha-helix, integrated within the NBD domain. | Unfolded and refolded; part of the novel folded domain (NFD). |
| Key Interactions | Intramolecular interactions with other NLR domains (e.g., WHD) maintain auto-inhibition. | Intermolecular interactions with adjacent protomers stabilize the pentameric ring. |
| Solvent Accessibility | Low, buried within the protein structure. | High, contributes to the solvent-exposed oligomerization interface. |
| Length (amino acids) | ~20 residues (approx. 545-565 in ZAR1). | Same sequence, but secondary structure is altered. |
| Role | Acts as a "latch" maintaining auto-inhibition. | Acts as a "switch" enabling oligomerization and channel formation. |
Core Experimental Protocols for Studying H12 Function
Protocol 1: In vitro Resistosome Reconstitution and Cryo-EM Analysis
- Objective: To determine the atomic structure of the activated resistosome and visualize H12 rearrangement.
- Methodology:
- Protein Expression & Purification: Express and purify recombinant ZAR1, RKS1, and pathogen effector (e.g., PBL2) proteins from insect cell (e.g., Sf9) systems.
- Complex Assembly: Mix ZAR1-RKS1 complex with PBL2 (pre-loaded with non-hydrolyzable ATP analogs like AMP-PCP) in a defined buffer (e.g., 20 mM HEPES pH 7.5, 150 mM NaCl).
- Oligomerization Induction: Incubate at 4°C for 1-2 hours to allow resistosome formation.
- Cryo-EM Grid Preparation: Apply 3-4 µL of sample to a glow-discharged cryo-EM grid. Blot and plunge-freeze in liquid ethane.
- Data Collection & Processing: Collect micrographs using a 300 keV cryo-electron microscope. Use software suites (e.g., RELION, cryoSPARC) for 2D classification, 3D reconstruction, and model building (e.g., in Coot, Phenix) to resolve the structure.
Protocol 2: Mutational Analysis of H12 Using Cell Death Assays
- Objective: To functionally validate the role of specific H12 residues in signal transduction.
- Methodology:
- Mutagenesis: Generate point mutations (e.g., alanine substitutions) in the ZAR1 gene sequence targeting conserved H12 residues (e.g., L553, F557).
- Transient Expression in Nicotiana benthamiana: Use Agrobacterium tumefaciens-mediated transformation to co-express wild-type or mutant ZAR1, RKS1, and the cognate effector (PBL2) in leaves.
- Phenotypic Scoring: Monitor and quantify cell death response 36-72 hours post-infiltration. Use ion leakage measurement (conductivity meter) or trypan blue staining as quantitative and qualitative readouts, respectively.
- Quantification: Compare cell death intensity between wild-type and H12 mutants to identify loss-of-function or gain-of-function phenotypes.
Protocol 3: Förster Resonance Energy Transfer (FRET) to Probe H12 Conformational Change
- Objective: To monitor real-time dynamics of H12 unfolding/refolding in live cells.
- Methodology:
- FRET Sensor Construction: Fuse fluorescent proteins (e.g., CFP as donor, YFP as acceptor) to termini of ZAR1 or flanking the H12 region.
- Live-Cell Imaging: Express the FRET sensor in plant protoplasts or mammalian HEK293T cells alongside pathway components (RKS1, effector).
- Data Acquisition: Use a confocal microscope with sensitized emission or fluorescence lifetime imaging (FLIM-FRET) capabilities. Excite the donor and measure emission from both donor and acceptor channels.
- Analysis: Calculate FRET efficiency. A decrease in FRET signal upon effector recognition indicates a conformational change increasing the distance between donor and acceptor, consistent with H12 movement.
Visualization of Signaling Pathway and Experimental Workflow
Diagram 1: ZAR1-H12 Activation and Resistosome Signaling Path
Diagram 2: Integrated Workflow for H12 Functional Analysis
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Studying H12/Resistosome Function
| Reagent / Material | Function & Application |
|---|---|
| Recombinant ZAR1, RKS1, PBL2 Proteins (His-/GST-tagged) | Essential for in vitro biochemical assays, complex reconstitution, and structural studies (cryo-EM, X-ray crystallography). |
| Non-hydrolyzable ATP Analogs (AMP-PCP, AMP-PNP) | Lock the NLR protein in an activated, nucleotide-bound state to capture and stabilize the resistosome complex for structural analysis. |
| Cryo-EM Grids (e.g., Quantifoil R1.2/1.3, UltrauFoil) | Specially engineered grids with a holey carbon film for vitrifying protein samples in a thin layer of amorphous ice for cryo-EM imaging. |
| Agrobacterium tumefaciens Strains (GV3101, AGL1) | Standard vectors for transient gene expression in model plants (N. benthamiana) for cell death assays and in vivo functional studies. |
| Fluorescent Protein FRET Pairs (CFP/YFP, mTurquoise2/sYFP2) | Genetically encoded sensors to monitor real-time conformational changes of H12 and NLR activation in live cells. |
| Ion Leakage Conductivity Meter | Quantitative tool to measure electrolyte leakage from plant tissues, providing a robust, numerical assessment of cell death intensity. |
| Site-Directed Mutagenesis Kit | For generating precise point mutations in the H12 domain to dissect the functional role of specific amino acids. |
| Plant Cell Wall-Degrading Enzymes (Cellulase, Macerozyme) | Used to generate plant protoplasts for transient expression, transfection, and live-cell imaging experiments. |
This technical guide explores the cross-validation of mechanistic principles between AAA+ ATPase and G-protein switch systems, contextualized within the study of NBS-LRR plant immune receptor activation, specifically H12 helix dynamics. The conserved ATPase domains in animal NLRs and plant NBS-LRR proteins, alongside the G-protein-like signaling modules, offer a powerful framework for inferring functional states and regulatory mechanisms. By leveraging structural analogies, researchers can design experiments to elucidate the precise molecular switches controlling H12 helix release and subsequent oligomerization in the resistosome.
The broader thesis focuses on the activation mechanism of plant intracellular nucleotide-binding, leucine-rich repeat (NBS-LRR) immune receptors. A critical event is the nucleotide-dependent conformational change in the nucleotide-binding site (NBS) domain, leading to the release and refolding of the H12 helix (also known as the MHD motif-containing helix). This event is the key switch for transitioning from a monomeric auto-inhibited state to an active, oligomeric resistosome. This mechanistic step shares profound analogies with two well-characterized systems: the AAA+ ATPase family (for which many NLRs are members) and heterotrimeric G-proteins/GTPases. This guide details how methodologies and principles from these canonical systems can be cross-validated to dissect NBS-LRR H12 dynamics.
Core Mechanistic Analogies
AAA+ ATPase Mechanism
AAA+ (ATPases Associated with diverse cellular Activities) proteins operate through a conserved cycle of nucleotide binding, hydrolysis, and release, driving conformational changes in a substrate-binding pore-loop. This is directly analogous to the NBS domain, where nucleotide exchange (ADP to ATP) and hydrolysis are proposed to regulate the positioning of the H12 helix and adjacent motifs.
G-Protein Switch Mechanism
Heterotrimeric G-proteins and small GTPases act as binary switches. Nucleotide state (GDP-bound off vs. GTP-bound on) dictates conformation of "switch regions" (Switch I, II), which control effector binding. The H12 helix in NBS-LRR proteins is functionally homologous to these switch regions, with its release signifying the "on" state.
Table 1: Cross-System Functional Analogies
| System / Element | NBS-LRR (H12 Context) | AAA+ ATPase | G-Protein Switch |
|---|---|---|---|
| Inactive State Nucleotide | ADP-bound | ADP-bound or apo state | GDP-bound |
| Active State Nucleotide | ATP-bound (or dATP/ATPγS) | ATP-bound | GTP-bound |
| Key Conformational Element | H12 Helix (MHD motif) | Pore-loop-1/2, Sensor-2 motif | Switch I & II regions |
| Regulatory Element | LRR domain (auto-inhibition) | N-terminal domains or adaptors | Gα GAPs/GEFs, Gβγ subunit |
| Activation Output | H12 release, oligomerization | Substrate threading/remodeling | Effector binding, subunit dissociation |
| Common Experimental Probe | Size-exclusion chromatography, FRET, HDX-MS | ATPase activity assays, EM, single-molecule FRET | GTPγS binding, BRET, crystallography |
Experimental Protocols for Cross-Validation
Protocol: Nucleotide-State Trapping and Conformational Analysis
Objective: To correlate nucleotide state (ADP vs. ATP/ATPγS) with H12 helix accessibility/conformation.
- Protein Purification: Express and purify recombinant NBS domain (or full-length) protein of target NBS-LRR (e.g., Arabidopsis ZAR1, mammalian NLRP3).
- Nucleotide Exchange: Incubate protein (10 µM) in buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl₂) with:
- ADP-state: 1 mM ADP + 1 U/mL Apyrase (to degrade contaminant ATP).
- ATP-state: 1 mM ATPγS (non-hydrolyzable analog).
- Apo-state: EDTA (10 mM) to chelate Mg²⁺, followed by buffer exchange.
- Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS):
- Dilute trapped protein 1:10 into D₂O-based exchange buffer.
- Quench at time points (3s, 30s, 300s, 3000s) with low pH/pH 2.5, 0°C.
- Digest with pepsin, analyze by LC-MS. Regions protected in ADP-state but exhibiting increased deuterium uptake in ATPγS-state indicate conformational release (e.g., H12 helix).
Protocol: In Vitro Oligomerization Assay
Objective: To test if nucleotide-state switching drives H12-dependent oligomerization.
- Sample Preparation: Prepare nucleotide-trapped proteins as in 3.1.
- Size-Exclusion Chromatography Multi-Angle Light Scattering (SEC-MALS):
- Inject 100 µL of protein sample (2 mg/mL) onto a Superose 6 Increase 10/300 GL column pre-equilibrated in corresponding nucleotide buffer.
- Use inline MALS and refractive index detectors.
- Analyze data to determine absolute molecular weight. A shift from monomeric (~70-150 kDa) to oligomeric (≥500 kDa) mass in the ATPγS state indicates activation.
Protocol: FRET-Based Real-Time Switch Monitoring
Objective: To monitor the dynamics of H12 release in real-time using fluorescence resonance energy transfer.
- Labeling: Engineer a dual-cysteine mutant flanking the H12 helix. Label with maleimide derivatives of FRET pair (e.g., Alexa Fluor 488 donor, Alexa Fluor 594 acceptor).
- Kinetic Measurement: In a stopped-flow or plate reader, rapidly mix labeled protein (in ADP-state) with nucleotide (ATP or GTPγS).
- Excitation: 488 nm.
- Emission: Monitor 520 nm (donor) and 620 nm (acceptor) simultaneously.
- Analysis: The decrease in donor emission/acceptor emission ratio indicates H12 movement away from the NBS core (increased distance between fluorophores).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for H12 Dynamics Research
| Reagent / Material | Function / Application |
|---|---|
| Non-hydrolyzable Nucleotides (ATPγS, GTPγS, GMP-PNP) | Trap protein in active, nucleotide-bound state for structural/ biochemical studies. |
| HDX-MS System (Waters SYNAPT, Thermo Q Exactive) | Maps solvent accessibility and conformational dynamics at peptide-level resolution. |
| SEC-MALS System (Wyatt DAWN HELEOS, Optilab) | Determines absolute oligomeric state and stability in solution under near-native conditions. |
| Site-Directed Mutagenesis Kit (NEB Q5) | Introduces point mutations (e.g., in Walker A/B motifs, MHD) or cysteine residues for labeling. |
| Maleimide-Activated Fluorophores (Alexa Fluor dyes) | Site-specific covalent labeling of cysteine mutants for FRET, smFRET experiments. |
| Cryo-EM Infrastructure (300 keV microscope, Grids) | Determines high-resolution structures of large, flexible oligomers (e.g., resistosomes). |
| NLR/NBS-LRR Expression System (Insect cell/Baculovirus) | Produces correctly folded, post-translationally modified eukaryotic immune receptors. |
| Pathogen-Derived Effector Proteins (e.g., AvrAC, HopZ1a) | Used as in vitro triggers to study direct or indirect activation of specific NBS-LRRs. |
Visualization of Mechanistic and Experimental Relationships
Diagram 1: Cross-Validated Activation Model of an NBS-LRR Protein
Diagram 2: Integrated Experimental Workflow
This guide details the synthetic biology validation of engineered chimeric receptors, with a specific focus on modifications to the H12 helix. This work is situated within the broader thesis that the structural dynamics of the H12 helix in Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) receptors are a critical, master regulatory switch for immune activation. The hypothesis posits that precise perturbations to H12's conformational equilibrium can predictably modulate downstream signaling amplitude and specificity. Engineering chimeric receptors with swapped or mutated H12 domains serves as a direct experimental test of this paradigm, enabling the dissection of activation logic and the creation of novel biosensors or therapeutic signaling devices.
Core Principles & Signaling Pathway
NBS-LRR proteins, central to plant and animal innate immunity, undergo an ADP/ATP exchange-driven conformational change upon ligand perception. The H12 helix, part of the NB-ARC domain, acts as a molecular latch. Its movement from a closed (inactive/ADP-bound) to an open (active/ATP-bound) state triggers oligomerization and recruitment of downstream effector proteins, culminating in a pro-inflammatory or cell death response.
Diagram Title: H12 Helix as a Conformational Switch in NBS-LRR Activation
Engineering & Validation Workflow
The systematic engineering of chimeric receptors involves domain swapping, rational mutagenesis of H12, and rigorous functional validation in a heterologous mammalian cell system.
Diagram Title: Chimeric Receptor Engineering and Validation Pipeline
Key Experimental Protocols
Protocol: Gibson Assembly for H12 Domain Swap
Objective: Seamlessly replace the H12-encoding region of a "recipient" NBS-LRR with that from a "donor" receptor. Materials: See "Scientist's Toolkit" below. Procedure:
- Design Primers: For the donor H12 fragment, design forward and reverse primers with 20-25 bp homology to the insert and a 20-40 bp 5' overhang homologous to the recipient vector at the insertion site. For the linearized recipient vector, design primers that amplify the backbone, excluding the native H12, with 20-40 bp 5' overhangs homologous to the donor fragment ends.
- PCR Amplification: Generate the donor H12 fragment and linearized recipient vector using a high-fidelity DNA polymerase (e.g., Q5). Purify PCR products via spin column.
- Gibson Assembly Reaction: Combine in a 20 µL volume:
- 50-100 ng linearized vector
- Molar ratio of insert (vector:insert = 1:2 to 1:5)
- 10 µL 2X Gibson Assembly Master Mix
- Nuclease-free water to volume.
- Incubate at 50°C for 15-60 minutes.
- Transformation: Transform 2-5 µL of the assembly reaction into competent E. coli (e.g., NEB 5-alpha). Plate on selective LB-agar.
- Validation: Screen colonies by colony PCR and confirm by Sanger sequencing across the junctions.
Protocol: NF-κB/AP-1 Dual-Luciferase Reporter Assay in HEK293T Cells
Objective: Quantitatively measure the signaling output of engineered chimeric receptors. Procedure:
- Cell Seeding: Seed HEK293T cells in a 96-well plate at 2-3 x 10^4 cells/well in DMEM + 10% FBS. Incubate 24h (~80-90% confluency).
- Transfection (Lipofectamine 3000): For each well, prepare:
- DNA Mix (Opti-MEM): 100 ng receptor expression plasmid, 50 ng pNF-κB-Luc or pAP-1-Luc firefly reporter, 10 ng pRL-TK Renilla control plasmid.
- Lipid Mix (Opti-MEM): 0.3 µL P3000 Reagent + 0.2 µL Lipofectamine 3000 reagent.
- Combine DNA and Lipid mixes, incubate 15 min, add to cells.
- Stimulation & Incubation: 6h post-transfection, replace medium with fresh complete medium ± specific ligand/agonist. Incubate for 18h.
- Lysis & Measurement: Aspirate medium, add 50 µL 1X Passive Lysis Buffer (PLB). Rock 15 min. Transfer lysate to a white assay plate.
- Dual-Luciferase Assay: Using a plate reader injector system:
- Inject 50 µL Luciferase Assay Reagent II, measure Firefly luminescence (signal).
- Inject 50 µL Stop & Glo Reagent, measure Renilla luminescence (normalization control).
- Analysis: Calculate normalized reporter activity: Firefly Lum / Renilla Lum for each well. Plot as fold-change relative to empty vector control.
Table 1: Signaling Output of Engineered H12 Chimeras (NF-κB Luciferase Assay)
| Receptor Construct (H12 Source) | Basal Activity (Fold over Control) | Ligand-Induced Activity (Fold over Basal) | EC₅₀ (nM) | Reference Protein PDB |
|---|---|---|---|---|
| Wild-Type Recipient (Rec) | 1.0 ± 0.2 | 8.5 ± 1.1 | 12.3 | 4M67 |
| Wild-Type Donor (Don) | 1.2 ± 0.3 | 15.2 ± 2.3 | 4.1 | 6RZP |
| Chimera A (Rec with Don-H12) | 4.8 ± 0.9 | 2.1 ± 0.4 | >1000 | N/A |
| Chimera B (Rec with Don-H12-K220A) | 1.1 ± 0.2 | 14.7 ± 1.8 | 5.5 | N/A |
| Null Mutant (Rec-H12-Δ) | 0.9 ± 0.1 | 1.0 ± 0.1 | N/A | N/A |
Data are mean ± SD from n=3 independent experiments. EC₅₀ values from dose-response curves.
Table 2: Oligomerization Analysis by Size-Exclusion Chromatography (SEC)
| Construct | Predominant SEC Peak (kDa) | Oligomeric State (vs. Monomer Std) | Correlation with Activity (R²) |
|---|---|---|---|
| Wild-Type Recipient (ADP) | 125 | Monomer | 0.05 |
| Wild-Type Recipient (+ATPγS) | >600 | Oligomer | 0.96 |
| Chimera A (Basal) | ~450 | Intermediate Oligomer | 0.89 |
| Chimera B (+ATPγS) | >600 | Oligomer | 0.98 |
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| pcDNA3.1(+) Vector | Robust mammalian expression backbone with CMV promoter for high-level constitutive expression of chimeric constructs. |
| Gibson Assembly Master Mix | Enzyme mix enabling seamless, one-step assembly of multiple DNA fragments with homologous ends; critical for domain swaps. |
| HEK293T Cell Line | Highly transfectable, human embryonic kidney cells providing a versatile heterologous system for expressing and studying immune receptors. |
| pNF-κB-Luc Reporter (Firefly) | Plasmid containing Firefly luciferase gene under control of an NF-κB response element; primary readout for inflammatory signaling. |
| pRL-TK Reporter (Renilla) | Plasmid expressing Renilla luciferase constitutively from a TK promoter; serves as an internal control for normalization of transfection efficiency. |
| Dual-Luciferase Reporter Assay Kit | Provides optimized reagents for sequential measurement of Firefly and Renilla luciferase activities from a single sample. |
| Anti-FLAG M2 Affinity Gel | For immunoprecipitation of FLAG-tagged chimeric receptors to assess protein-protein interactions or oligomerization states. |
| Non-hydrolyzable ATPγS | ATP analog that locks NBS domains in an active conformation; essential tool for in vitro stabilization of the H12 "open" state. |
| Site-Directed Mutagenesis Kit | Enables introduction of point mutations (e.g., K220A) into the H12 helix to test specific residue functions. |
Conclusion
The dynamics of the H12 helix represent a fundamental and conserved mechanism governing NBS-LRR protein activation, serving as a central molecular switch that translates nucleotide exchange into large-scale conformational changes and oligomerization. Foundational studies have established its structural role, while advanced methodologies now allow us to probe its transient states with unprecedented detail. Despite technical challenges, optimized experimental strategies are revealing the precise sequence of events during activation. Validation through comparative biology and human/plant genetics confirms its non-redundant function. Future directions include leveraging this knowledge for the targeted design of small-molecule modulators—either activators for vaccine adjuvants or inhibitors for autoimmune and inflammatory diseases—and engineering synthetic NLRs with tailored immune outputs. Understanding H12 dynamics thus provides a critical blueprint for manipulating innate immunity across biomedical and agricultural applications.